Alarmins support the pathogenesis of chronic respiratory diseases as asthma through ferroptosis and the axis composed of high mobility group box 1 (HMGB1) and interleukin (IL)-33. T and IL-33. We describe the mechanisms supporting the development and progression of chronic respiratory diseases and the role of HMBG1 and IL-33 are described.
- respiratory diseases
- alarmins
- high mobility group box 1
- interleukin-33
- microRNAs
1. Introduction
1.1. Chronic Respiratory Diseases
1.2. General Considerations Regarding High Mobility Group Box 1, Interleukin-33 and microRNAs
1.2.1. High Mobility Group Box 1
HMGB1 is a chromatin-binding protein that weakly binds DNA. It is formed by 215 amino acid residues divided into three different parts: two tandem high mobility group box domains separated by a short linker and an acidic C-terminal tail. It binds DNA to modify the chromatin configuration while preserving genome stability [6]. HMGB1 produces numerous effects inside the nucleus. It helps nucleosome development, operates as a DNA chaperone to support DNA duplication and repair [7][8][7,8], and is doubtlessly implicated in the pathogenesis of tumours [9][10][9,10]. It is also involved in V(D)J transcription and stimulates autophagy and cell survival when it occurs. It is produced and released by macrophages, natural killer (NK) cells, and dendritic cells (DCs) during inflammation, or by cells after necrosis or cell death. HMGB1 is an alarmin and, thus, causes reactions of both adaptive and innate immune effectors. It operates via the receptor for advanced glycation end products (RAGE) and other receptors, such as Toll-like receptors (TLRs) 2 and 4 [11]. HMGB1 works as a damage-associated molecular pattern (DAMP) when discharged from injured cells or released from stimulated macrophages, triggering numerous ligands [12][13][14][15][16][17][12,13,14,15,16,17]. This alarmin may be involved in other processes, such as coagulation. HMGB1 intensifies the sterile inflammation correlated with infection and thrombosis [18]. Platelets accumulate HMGB1, transfer it to the cell surface when stimulated and then discharge it into microparticles. Moreover, thrombocytes can connect to HMGB1 via the RAGE, coherent with possible autocrine effects [19][20][21][19,20,21].1.2.2. Interleukin-33
IL-33 is a 30-KDa protein belonging to the IL-1 cytokine family. Its structure presents two different domains: the C-terminal and the N-terminal domain. The C-terminal domain is responsible for extracellular ST2-dependent activity. The N-terminal domain matches IL-33 to the nucleus and has a transcriptional repressor effect [22]. The IL-33 chromatin-binding motif present in this domain facilitates attachment to histone dimers and changes the chromatin configuration. This mechanism is implicated in the transcription of genes, altering gene repression [8]. The active form of IL-33 does not hold a signal peptide, so it is not discharged via a usual secretory pathway; rather, this cytokine is discharged when cells are injured and works like an alarmin [23]. IL-33 is released by epithelial and endothelial cells but can be released into the airways by other cells, such as DCs and macrophages [24][25][24,25]. IL-33 is an essential controller of immune responses operating on DCs, mast cells, group 2 innate lymphoid cells (ILC2s), macrophages, and CD4 + T2 cells, provoking a type 2 immune response [5]. Moreover, this cytokine can stimulate the ST2 of Tregs, demonstrating its capability to reduce inflammation. However, IL-33 can stimulate the synthesis of IL-8 via the stimulation of the JNK/c-Jun/AP-1 pathway, provoking inflammatory conditions. The control of gene expression is also due to the silencing of RNAs. IL-33 has been shown to silence Rip2 by sequentially reducing the expression of mRNAs associated with factors such as IL-17, iNOS, E-selectin, ICAM-1, VCAM-1, and TSLP. Furthermore, NOD-like receptor protein 3 (NLRP3) gene silencing reduces IL-33 synthesis [26].1.2.3. microRNAs
Modifications in gene expression can be caused by numerous forms of non-coding RNA, including long non-coding RNA (lncRNA), circular RNA (circRNA), and microRNA (miRNA). MicroRNAs are RNA sequences of 17–24 nucleotides that modify gene expression by connecting to messenger RNA (mRNA) transcripts in the 3′-untranslated region or coding sequence, causing a reduction in mRNA translation and protein production, and stimulating mRNA elimination [27][28][29,30]. However, connecting miRNAs to mRNA transcripts may be an inaccurate process, involving chains of only 6–8 nucleotides; this implies that each miRNA can have several different mRNA targets and, hypothetically, can affect numerous genes and different functional systems [29][30][31,32]. Furthermore, miRNAs can be categorized into isomiRs, which contain modifications in the sequence dimension or nucleotides at the 3′ or 5′ ends, occasionally with alterations in activity and targets [31][33]. It has been shown that although miRNAs are present and operate within the cytoplasm, they can also be discharged into vesicles called exosomes. These vesicles arrive at the circulatory system, where they may facilitate cellular interactions [32][34]. MiRNA profile changes and altered expression compared to healthy subjects can be observed in different diseases. These variations in miRNA profiles promote some genes and reduce the expression of others [29][31].2. Alarmins and Chronic Respiratory Diseases
2.1. Asthma and Alarmins
Asthma is a chronic pathology characterized by airway inflammation, hyperresponsiveness, submucosal fibrosis and increased mucus production [33][35]. It affects more than 270 million people, affecting both children and adults. Approximately 3–10% of all patients present with a severe form of the disease [34][36]. Asthma is distinguished by several immunological alterations provoked by the relationship between the environment and the patient’s genetic substrate. Based on immunological alterations, the disease can be classified into T2 asthma and non-T2 asthma [35][37]. This classification depends on the concentrations of type 2 cytokines [36][38]. The pathogenesis of asthma is due to type 2-mediated allergic inflammation that induces barrier defences at the mucosal surfaces and stimulates the generation of type 2 cytokines, including IL-4, IL-5, IL-9, and IL-13, and the shift of antibodies to IgE [37][38][39][39,40,41]. An innate type 2 immune response requires CD4+ T cells but depends on ILC2s [40][41][42][42,43,44]. ILC2s were initially recognized as cells not derived from the bone marrow and spleen. These are IL-25-dependent non-T non-B cells that were identified in the lung, spleen, liver, mesenteric lymph node, and peritoneum [43][44][45,46]. ILC2s induce T-helper cell (Th)2-related lung inflammation by controlling the delivery of Th2 cytokines. IL-33 plus IL-2, IL-7, IL-25, and TSLP stimulate lung ILC2s by reacting to allergen-induced tissue injury [45][46][47,48]. Lung ILC2s induce the production of a significant amount of IL-5 and IL-13 but not IL-4. In any case, ILC2 stimulation is mainly due to IL-33, which is induced in type 2 pneumocytes after contact with allergens [47][48][49][50][51][49,50,51,52,53]. It is noteworthy that different asthma types may respond to various treatment regimens [52][53][54,55]. A different approach to show the correlation between IL-33 and HMGB1 evaluated whether the P2Y13 receptor (P2Y13-R), a purinergic G protein-coupled receptor (GPCR), controlled the discharge of IL-33 and HMGB1 [54][65]. Human and mouse airway epithelial cells and C57Bl/6 mice were treated with different aeroallergens or respiratory viruses. The discharge and transfer of alarmins from the nucleus to the cytoplasm were evaluated. The effect of the P2Y13-R on the activity of airway epithelial cells was evaluated during and after experimental asthma by employing antagonists or animals with P2Y13-R gene deletion. Allergen contact provoked the discharge of ADP and ATP, molecules that stimulate P2Y13-R. Allergens, ATP, ADP, or contact with viruses caused the transfer of IL-33 and HMGB1 from the nucleus to the cytoplasm and their subsequent release; this effect was annulled by genetic deletion or the use of P2Y13 antagonists [54][65]. The function of alarmins in some pathological processes, such as ferroptosis, supports the existence of an axis composed of HMGB1 and IL-33. A link between IL-33 and HMGB1 in neutrophilic asthma has been evidenced and could be represented by ferroptosis. Ferroptosis is a type of regulated cell death (RCD) system that is dependent on reactive oxygen species (ROS) [55][67]. HMGB1 is discharged into the extracellular area in numerous forms of RCD and is correlated to the genesis of inflammation, both due to infections and aseptic. As an alarmin, HMGB1 could provoke the discharge of inflammatory substances, such as cytokines, after ferroptosis initiation. It has been reported that ferroptosis could significantly stimulate inflammation through IL-33 delivery [56][76]. The reduction in IL-33 decreases the inflammation caused by house dust mites (HDM), including eosinophil and neutrophil infiltration [57][77].2.2. Alarmins and Respiratory Syncytial Virus Infection
Interesting data on the role of HMGB1 and IL-33 have also emerged in the context of different respiratory diseases, such as disorders derived from respiratory syncytial virus (RSV) infection and cystic fibrosis (CF). RSV is a rapidly transmissible virus that provokes acute lung infections in children. The signs of RSV infection are slight; nevertheless, a subset of patients presents with severe RSV-correlated bronchiolitis. Inflammation provoked by RSV infection can include both the superior zone with nasal inflammation and rhinorrhoea and the inferior respiratory area, resulting in wheezing, bronchiolitis, and coughing [58][59][80,81]. Severe RSV-induced bronchiolitis causes necrosis, oedema, the shedding of airway epithelial cells, mucus overproduction and peribronchiolar inflammation. These elements cause airway obstruction [60][82]. Generally, RSV induces a type 1 immune response. Nevertheless, type 2 cytokines may also be released after RSV infection. There are increasing suggestions that children with critical RSV-correlated bronchiolitis are at a higher risk of presenting with asthma throughout their lives [61][62][63][64][83,84,85,86]. The epithelial-originated cytokines IL-33 and IL-25 and the alarmin HMGB1 stimulate and control ILC2 activity, driving the development of type 2-provoked pulmonary pathologies [65][66][67][87,88,89]. The management of RSV-infected airway epithelial cells in a culture with a xanthine oxidase inhibitor reduced the generation of both IL-33 and TSLP [68][93]. In vivo, the dispensation of a xanthine oxidase inhibitor reduced the generation of bronchoalveolar lavage fluid (BALF) IL-33 and decreased the number of ILC2s in the lungs of mice [68][93]. The role played by the IL-33/HMGB1 axis in respiratory infections has been confirmed in other experimental studies. IFN-beta promoter stimulator I (IPS-1) deficit influences susceptibility to viral bronchiolitis and provokes the onset of type 2 inflammation and airway alterations. The main reason for this condition is a deficiency in antiviral cytokine generation that can cause a significantly higher viral burden in epithelial cells, which die due to necrosis, and the production of IL-33 and HMGB1. This condition is associated with the increased generation of ILC2 type 2 cytokines, such as IL-5 and IL-13, and airway smooth muscle modification. Significantly, this type 2 inflammatory condition does not occur in adult animals with an IPS-1 deficit affected by infection, indicating that this condition is dependent on age and reflecting the human distribution of sensitivity to RSV bronchiolitis. The appearance of a type 2 immune response after an acute and severe alteration of epithelial function is coherent with its fundamental function in regulating lesion repair [69][70][94,95] as ILC2s, which are the principal ILC component in the lungs, are the main controllers of tissue damage repair [71][72][96,97].2.3. Alarmins and Cystic Fibrosis
In CF, recurrent lung inflammation provokes increasing tissue injury. After cell damage and the onset of inflammation, necrotic cells discharge proteins as danger signals, which participate in the eradication of pathogens and stimulate cell repair [73][103]. However, the excessive liberation of such substances may increase inflammation, accelerating tissue alterations. Considering the Th17/Th2 form of pulmonary phlogosis in CF patients and its correlation to CF-related pathogens, IL-33 and HMGB1 may participate in inflammation. Unlike the diffuse production of HMGB1, IL-33 is basically produced in epithelial cells [73][103] and has a role in tissue inflammation correlated to pathogen infection [74][75][76][77][104,105,106,107]. To evaluate the influence of IL-33 and HMGB1 in CF inflammation, researchers considered the IL-33 concentrations in the BALF of CF patients and evaluated subjects with frequent pulmonary infections. The IL-33 concentrations were significantly higher in CF subjects than in non-CF subjects, while the HMGB1 concentrations were higher in non-CF controls with repeated infections [78][108]. Furthermore, the Th2 cytokine pattern in CF patients has been correlated to a deficit of IFN-c [79][110], possibly due to an effect of IL-33. Moreover, IL-33 regulates neutrophil inflow, a specific aspect of CF. In addition, contact with Pseudomonas aeruginosa has been reported to stimulate IL-33 but not HMGB1 expression, and this finding might correlate this infection with an increased Th2 reaction [80][111]. Finally, other relationships between Il-33 and CF have been observed, such as an association between IL-33 expression and the lack of a CF transmembrane conductance regulator [80][111].3. MicroRNAs and Chronic Respiratory Diseases
3.1. MicroRNAs and Asthma
The role played by miRNAs in respiratory diseases is closely linked to the specific miRNA, as the non-coding genetic material can play a triggering or protective role. MiRNAs are involved in the genesis of asthma, as a relevant modification in the miRNA generation of airway cells has been described by numerous studies [81][82][83][84][85][86][113,114,115,116,117,118]. An analysis stated that epithelial miRNA-141 controlled the presence of airway mucus in asthma [86][118], while different studies implicated epithelial miRNAs in airway eosinophilia; moreover, an evaluation of miRNAs in asthma stated that a group of epithelial miRNAs was reduced in these subjects [86][118]. For instance, miRNA-30a-3pa, an miRNA also implicated in the growth and apoptosis of tumour cells [87][88][119,120], was considerably reduced in the blood of asthmatic subjects and is probably involved in airway eosinophilic inflammation. Moreover, it was reported that miRNA-3934 concentrations might be employed to differentiate asthma subjects from non-asthmatic subjects. Regarding the mechanism, miRNA-3934 suppressed the increase in AGEs, which provoked an increase in apoptosis in basophils [89][123]. These findings suggest that miRNA-3934 can reduce the onset of asthma by targeting the RAGE and probably inhibiting other pathways, such as the TGF-β/Smad signalling pathway. Even more interesting are the data relating to a possible correlation between miRNAs and HMGB1 in the pathogenesis of asthmatic disease. There are three runt-related transcriptional factor (RUNX) genes, namely RUNX1, RUNX2, and RUNX3, as well as maternal smoking, which have been reported to potentially support the onset of asthma in children by increasing the expression of RUNX1 [90][132]. Moreover, RUNX2 is described as stimulating the gene transcription of a sterile alpha motif (SAM) pointed domain comprising an ETS transcription factor (SPDEF) and may join the promoter of HMGB1 [91][92][133,134]. Wu et al. evaluated miRNA-30a-3p production in asthma patients and non-asthmatic subjects and considered the relationship between miRNA-30a-3p and airway eosinophilia [93][135]. They found that miRNA-30a-3p production was significantly reduced in the bronchial brushings of asthma patients compared to normal controls. Epithelial miRNA-30a-3p production was negatively related to several factors indicating airway eosinophilia, such as eosinophils in sputum or bronchial biopsies and exhaled nitric oxide in patients. The scautholars demonstrated that RUNX2 is a target of miRNA-30a-3p and augments HMGB1 expression. HMGB1 and RUNX2 production are both increased in the airway epithelium and are related to each other in asthmatic subjects. A reduction in miRNA-30a-3p increased RUNX2 and HMGB1 production, while increasing RUNX2 stimulated HMGB1 in BEAS-2B cells. Interestingly, an airway increases in mmu-miRNA-30a-3p inhibited HMGB1 and RUNX2 expression and reduced airway eosinophilia in an experimental animal model [93][135]. Therefore, epithelial miRNA-30a-3p could target the RUNX2/HMGB1 axis to reduce airway eosinophilia in asthma (Figure 12).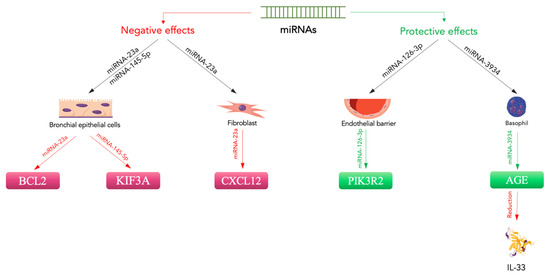
3.2. MicroRNAs and Acute Respiratory Distress Syndrome
ALI or its more critical manifestation, ARDS, is a severe acute pulmonary pathology with a significant mortality rate. ALI/ARDS is caused by different pulmonary conditions, such as pneumonia, or indirect extra-pulmonary damage, such as sepsis [97][136]. Its characteristics include alteration of the alveolar-capillary membrane, increased inflammation and reduced alveolar fluid clearance, with secondary hypoxemia, pulmonary oedema, and abnormal gas exchange [98][99][100][137,138,139].
MiRNAs and HMGB1 could also play a role in the genesis of ARDS. A report stated that the increased expression of miRNA-181b reduced the gene expression of importin-a3 and was able to decrease lung damage and mortality rates in ARDS animals [101][142]. Furthermore, Rao et al. showed that modification of the cytokine signalling suppressor 1 (SOCS1) in an animal model of ARDS reduced inflammatory cytokine production and inflammatory cell accumulation in miRNA-155 (−/−) animals compared to wild-type mice [102][143].
Finally, in different experiments, researchers assessed which miRNA could improve ARDS by targeting HMGB1 [103][144]. In experimental in vitro and in vivo models of LPS-induced ARDS models, they studied the effect of miRNA-574-5p on the production of HMGB1, pro-inflammatory cytokines and inflammasomes. MiRNA-574-5p seemed able to reduce the inflammatory reaction by operating on HMGB1. Stimulating the production of miRNA-574-5p or HMGB1 siRNA silencing reduced the stimulation of the NLRP3 inflammasome. Additionally, increased expression of HMGB1 overturned the anti-inflammatory action of miRNA-574-5p. In vivo, the increased production of miRNA-574-5p reduced interstitial oedema and alveolar leucocyte infiltration in ARDS animals [103][144].