1. T-Cells in IBD
Under steady state conditions, the gut contains scattered interepithelial lymphocytes and innate lymphocytes in the epithelial layer of the intestinal mucosa, with very few CD4 T-cells
[1][41]. In contrast, IBD is associated with an abundance of CD4 T-cells in the epithelial layer of the inflamed intestinal mucosa
[2][42] or with normal numbers of lamina propria and epithelial CD4 T-cells
[3][4][43,44] but showing increased activation
[5][6][7][45,46,47] and phenotypic alterations
[8][48].
T-cells release interleukin (IL)-2, which signals in an autocrine manner via the IL-2 receptor, whose α chain, called CD25, is expressed on T-cells upon antigen recognition and activation. IBD is characterized by elevated numbers of hiCD25+ cells, specifically affecting T-cells in CD and macrophages in UC
[9][49]. Some intestinal CD4 T-cells from CD patients, but not UC patients, also express high levels of the activating natural killer group 2D receptor (NKG2D)
[10][50], whose stimulation in combination with that of the TCR promotes the cytotoxic capacity of CD4 T-cells, plus the release of the pro-inflammatory cytokines tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and IL-17A
[10][11][50,51].
CD has usually been considered a type 1-driven disease, with the exacerbated production and activation of Th1 and Th17 cells and an elevated presence of their major cytokines IL-12, IL-23, IFN-γ, and IL-17. In contrast, UC has been designated as type 2-driven inflammation, linked to an elevated participation of Th2 and Th9 cells and their principal cytokines IL-13, IL-5, and IL-9
[12][13][52,53].
2. T Helper 1 (Th1) Cells
Th1 cells facilitate the eradication of intracellular pathogens, including parasites, protozoa, viruses, and intracellular bacteria, and intervene in cell-mediated immunity and delayed-type hypersensitivity reactions
[14][54]. Th1 cells release IFN-γ and TNF-α, which stimulate innate immune cells, such as neutrophils and macrophages, and non-immune cells, such as epithelial cells and fibroblasts
[15][16][55,56]. Th1 cells also release IFN-γ and IL-2 to recruit CD8 effector cytotoxic T-cells (CD8 CTL)
[17][57].
Upon antigen recognition and the activation of a naïve CD4 T-cell, Th1 differentiation is mediated by the binding of IL-12 produced by the cognate APC. IL-12 induces T-cell expression of the master Th1 transcription factor T-box-containing protein (T-bet), encoded by the gene TBX21, and the cytokine IFN-γ, in both cases through a process dependent on STAT4 signaling stimulation
[15][16][18][55,56,58]. T-bet increases the expression of IL-12 receptor subunit β2 (IL-12Rβ2), allowing synergistic IL-12 and STAT4 signaling to further increase IFN-γ generation
[19][20][21][26,59,60].
In intestinal homeostasis, Th1 cells can prevent pathogen invasion and pathogen-derived antigens from mediating intestinal inflammation. Beside their direct antibacterial action, Th1 cells also ameliorate intestinal inflammation by secreting IL-2 and IL-10 to promote Treg stimulation. Moreover, Th1 cells can facilitate intestinal stem cell (ISC) proliferation and intraepithelial cell self-restoration by releasing low concentrations of TNF-α. Th1 cells thus constitute an immune barrier indispensable for intestinal homeostasis
[22][61].
A pathogenic role for Th1 cells has been described in the course of IBD (
Figure 12). An excessive Th1 response has been observed in the inflamed mucosa and serum of IBD patients
[23][62]. Classically, an exacerbated Th1 response has been linked to CD, whereas UC has been considered a Th2 cell-driven disease
[24][63]. However, both UC and CD feature activated effector Th1 cells, suggesting that Th1 cells are implicated in the origin and development of mucosal inflammation in IBD
[25][64].
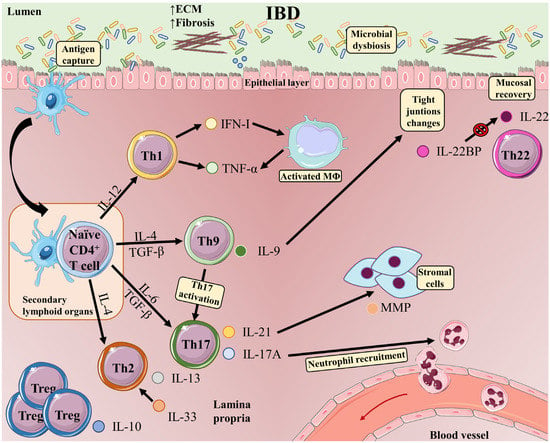
Figure 12. T-cell subsets and functions in the intestinal mucosa in inflammatory bowel disease. The development of IBD is induced by multiple phenomena occurring in the gastrointestinal tract: microbial dysbiosis, disruption of the mucus layer, dysregulation of epithelial tight junctions, defects in the number and function of Paneth cells, and increased intestinal permeability. These events massively increase bacterial exposure. In this context, antigen-bearing DCs capture antigens and migrate to secondary lymphoid organs, where they present antigens to naive T-cells. Once activated, CD4 T-cells undergo proliferation and differentiation into different effector T-cell subsets (Th1, Th9, Th17, and Th2 cells). Differentiated Th cells migrate back to the gut, where they carry out inflammatory functions, such as production of IFN-γ in the case of Th1 cells or IL-17A (which plays an important role in recruiting neutrophils to sites of active inflammation) and IL-21 (which induces MMP production by stromal cells) in the case of Th17 cells. Cytokines released by Th1 cells favor activation of macrophages, which release TNF-α and trigger epithelial-cell apoptosis. Th9 cells produce IL-9, which can act as a proinflammatory cytokine, activating Th17 cells. The presence of IL-9 is associated with alterations in the expression of tight junctions, and intestinal overproduction of IL-9 is likely to impair epithelial-barrier integrity and compromise tolerance to commensal bacteria, eventually progressing to inflammation. IL-33 is upregulated in UC patients and drives a Th2-like cytokine response. Elevated IL-33 production Th2 cells have also been reported in UC patients. Proinflammatory signals in IBD are counterbalanced by IL-10 produced by Tregs. IL-22 released by Th22 cells maintains intestinal epithelial barrier function. In inflamed intestinal tissue, CD4 T-cells are a major source of IL-22BP, which blocks IL-22 signaling.
3. T Helper 2 (Th2) Cells
Th2 cells participate in the elimination of extracellular microbes and intestinal helminths and support IgE-mediated B-cell responses by secreting IL-4, IL-5, IL-13, and IL-10
[26][90]. Th2 polarization is mediated by IL-4-ligation–dependent STAT6 signaling and the production of the Th2 master transcription factor GATA binding protein 3 (GATA-3)
[27][28][91,92]. In addition to IL-4, Th2 cells produce the cytokines IL-5, IL-13, IL-21, and IL-25. Th2 cytokines prevent Th1 differentiation and promote the activation of macrophages
[14][15][54,55]. Impaired Th2 responses are linked to allergies and asthma
[29][30][31][32][33][34][93,94,95,96,97,98].
Oxazolone-induced colitis in mice involves a Th2 response featuring IL-5 and IL-4 production
[35][99]. Another important Th2 cytokine is IL-33, which is elevated in UC patients and in mouse models of colitis induced with trinitrobenzenesulfonic acid (TNBS) or DSS. Moreover, IL-33 and the IL-33 receptor ST2 (suppression of tumorigenicity 2) are associated with IBD risk loci
[13][36][37][38][39][40][41][42][43][53,100,101,102,103,104,105,106,107]. A lack of ST2 in mice diminishes colitis, whereas the administration of exogenous IL-33 aggravates the condition. These effects are associated with increased amounts of the Th2 cytokines IL-4, IL-5, and IL-13; major reductions in IL-17 and IFN- γ; damage to the epithelial barrier; and delayed wound recovery in the damaged colonic epithelium
[13][36][37][38][41][42][43][53,100,101,102,105,106,107]. In contrast, IL-33 protects against intestinal inflammation by promoting the differentiation of forkhead box P3 (Foxp3)+ Tregs and innate lymphoid cells (ILCs) and by inducing the expression of amphiregulin
[44][45][108,109].
4. T Helper 9 (Th9) Cells
Th9 cells, like Th2 cells, intervene in the response to intestinal helminths
[46][113] and have been linked to allergy and autoimmunity
[47][114]. The differentiation of Th9 cells is induced by the concurrent action of IL-4 and transforming growth factor-beta (TGF-β). IL-4 binding to the IL-4 receptor triggers GATA3 transcription and the phosphorylation and dimerization of STAT6, promoting Th2 differentiation, whereas TGF-β activates FOXP3, inducing Treg differentiation
[48][49][115,116]. In combination, IL-4 and TGF-β induce the production of IL-9 and the polarization of CD4 T-cells towards the Th9 phenotype
[50][51][52][117,118,119]. Th9 differentiation depends on multiple transcription factors, including PU.1 and IRF4
[53][54][120,121]. Th9 differentiation can also be induced by other molecular combinations
[55][122], such as IL-4 plus IL-1β
[56][123]. Th9 cells are the main source of IL9, but also release IL-10
[51][57][118,124]. IL-9 can act as a proinflammatory cytokine, activating Th17 cells
[58][125], and shares the same γ-chain receptor as IL-4, IL-2, and IL-15. IL-9 binding to its receptor activates janus kinase (JAK)1 and JAK3, which form dimers with STAT3, STAT5, or STAT1
[59][60][61][126,127,128].
The contribution of Th9 cells and their role in gut immunity have been demonstrated in several studies. Altered tissue integrity and continuous inflammation during flare-up episodes in UC are associated with IL-9 release by Th9 cells in the colon
[62][63][129,130]. The presence of Th9-derived IL-9 is associated with alterations in the expression of tight junctions
[64][131].
5. T Helper 17 (Th17) Cells
Th17 cells protect the host from bacterial and fungal infections on mucosal surfaces but are also implicated in inflammatory and autoimmune diseases
[65][146]. Th17 cells have thus been identified as pathogenic cells in relation to tissue inflammation and autoimmune disease
[66][67][68][147,148,149]. However, it is becoming clear that Th17 cells also have a non-pathogenic phenotype with immune-modulatory functions
[22][69][70][71][72][61,150,151,152,153].
Pathogenic and non-pathogenic Th17 cells can be polarized in vitro
[73][154]. A combination of IL-6, IL-23, and IL-1β promotes pathogenic Th17 differentiation
[74][75][155,156], whereas TGF-β1, in addition to IL-6, favors non-pathogenic Th17 cells
[69][76][77][78][150,157,158,159]. IL-23 appears not to promote Th17 differentiation directly since naïve T-cells do not express the IL-23 receptor (IL-23R) in vitro, thus suggesting that IL-23 stabilizes the Th17 phenotype and promotes Th17 cell survival
[75][156].
Pathogenic and non-pathogenic Th17 cells both express the transcription factor retinoic acid receptor-related orphan nuclear receptor gamma (RORγt)
[79][160] in a STAT3-dependent manner
[80][161] and produce IL-17
[22][61]; however, they have distinct genetic signatures, one contributing to immune injury and the other to immune homeostasis
[73][81][154,162]. Pathogenic Th17 cells are characterized by the production of pro-inflammatory molecules, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-23R, and by a low expression of immune-regulatory molecules, such as IL-10 and CD5 molecule like (CD5L). In contrast, non-pathogenic Th17 cells produce low amounts of GM-CSF and IL-23R and high amounts of IL-10 and CD5L, facilitating tissue homeostasis
[69][70][75][82][83][150,151,156,163,164].
Th17 cells are more numerous in the peripheral blood of IBD patients, and several major Th17 cytokines, such as IL-17, IL-21, and IL-23, are abundant in the inflamed mucosa of these patients
[84][178].
IL-17 also modulates anti-microbial peptide release, potentially modulating microbial populations within the gut in IBD
[85][195]. IL-17, in concert with fibroblast growth factor 2, also controls both epithelial barrier maintenance and bacterial homeostasis in the intestine
[86][169]. Together, these data indicate a proinflammatory effect of Th17 in concert with a role in maintaining a healthy epithelial barrier and an optimal bacterial balance.
IL-23 promotes the expansion of pathogenic Th17 cells by maintaining Th17 signature genes, upregulating effector genes, such as IL17A, IL17F, or IL22, or repressing suppressive factors. Moreover, IL17 and IL23 signaling promote pro-inflammatory molecules such as TNF, IFNγ, IL22, lymphotoxin, and IL1β
[87][176]. Several mouse models of colitis have shown an augmented production of IL23
[88][89][90][91][197,198,199,200]. In patients, treatment with selective IL23 inhibitors promotes better response rates in the cohort of CD patients that failed prior anti-TNF therapy (reviewed in
[87][176]), and IL23 targeting in UC patient, is safe and effective and promote and sustain clinical remission, low inflammation, mucosal healing, and an improved quality of life (reviewed in
[92][201]). These experiments indicate the importance of the IL23/IL17 axis in mucosal inflammation.
6. T Helper 22 (Th22) Cells
Th22 cells protect against tissue damage and bacterial infection by producing the IL-10 family member IL-22
[93][94][95][202,203,204]. Th22 cells also produce IL-13, fibroblast growth factor, chemokines, and TNFα. IL-22 is also secreted by Th1 and Th17 cells, but Th22 cells are able to secrete IL-22 without producing IFN-γ or IL-17
[14][96][54,205]. IL-22 is also secreted by NKs, γδT cells, ILC3s, and some nonlymphoid cells
[97][206]. Th22 cells express the chemokine receptors CCR10, CCR6, and CCR4, and their differentiation is promoted by the activation of STAT3 and the aryl hydrocarbon receptor (AHR) by IL-6, TNF-α, and IL-1β and is diminished by TGF-β
[98][99][207,208].
IL-22 enhances innate immunity by modulating cell differentiation, chemokine secretion, and antimicrobial peptide (AMP) secretion
[100][101][102][209,210,211]. In the intestinal epithelium, IL-22 promotes the secretion of AMPs, such as β defensins and lipocalin 2 and the mucin proteins MUC1 and MUC3
[103][212]. IL-22 can also promote the secretion by human colonic myofibroblasts of the anti-inflammatory factor IL-11 and inflammatory molecules, such as IL-6 and CXCL chemokines
[104][213].
In healthy individuals, IL-22 is released mainly in the gastrointestinal tract, where it favors mucosal recovery
[105][106][214,215]. This beneficial effect is mediated by the binding of IL-22 to the receptor IL-22R, whose expression is mostly limited to epithelial cells
[105][214].
IL-22 maintains intestinal epithelial barrier function by promoting the release of antimicrobial peptides
[93][202] and mucins
[106][215], as well as by facilitating intestinal epithelial cell survival and proliferation
[105][214]. IL-22 can increase the production of anti-inflammatory factors, such as IL-11, that also protect epithelial barrier function
[107][216].
However, elevated levels of IL-22 can be detrimental
[97][206], enhancing the production of inflammatory mediators, such as IL-6 and CXCL chemokines by human colonic myofibroblasts
[108][217]. IL-22 modulates neutrophil recruitment to the colon by controlling the expression of neutrophil-active CXC-family chemokines in ulcerative colitis; by this mechanism, the augmented expression of IL-22 is associated with treatment resistance to an anti-IL-12/23 p40 subunit monoclonal antibody
[109][218].
IL-22 is secreted at low levels, and is mostly maintained in a biologically inactive state through the action of IL-22 binding protein (IL-22BP, also known as IL-22RA2), produced by intestinal DCs and macrophages in the gut lamina propria and secondary lymphoid structures
[110][111][112][113][114][219,220,221,222,223]. In inflamed intestinal tissue, the main producers of IL-22BP are CD4 T-cells
[108][111][217,220]. IL-22BP is a soluble receptor homolog that attaches to IL-22 with greater affinity than IL-22R, preventing IL-22 from binding to its receptor and thereby blocking IL-22 signaling
[115][116][224,225]. Elevated levels of IL-22 and IL-22BP mRNA and protein have been detected in inflamed tissue from CD and UC patients
[111][117][118][220,226,227]. Consistent with these findings, the IL-22-associated protection against DSS-induced colitis is increased in IL-22BP deficient rats
[119][228], and IL-22BP aggravates T-cell-mediated colitis in mice
[111][220]. IL-22BP expression is reduced in infectious colitis but not in inflamed tissues in IBD, indicating potential pathophysiological significance for IL-22BP-dependent alterations in IL-22 bioactivity
[111][119][220,228].These responses may vary between patients and differ according to the extent of histological damage. For example, CD patients with granuloma are reported to have increased frequencies of IL-22+ and IL-22+ IFN-γ+ cells in colonic tissue
[120][229].
7. Regulatory T-Cells (Treg)
Treg cells suppress immune responses and maintain peripheral tolerance and immune homeostasis
[121][231]. Tregs are divided into thymic-derived Tregs, also called natural Treg cells (nTregs)
[122][232], and post-thymic maturation peripheral Tregs (pTregs)
[123][124][125][126][127][233,234,235,236,237]. Tregs induced in vitro by the addition of TGF-β and IL-2 to naïve CD4 T-cells are called inducible Tregs (iTregs)
[127][128][237,238].
Tregs are characterized by the secretion of the inhibitory cytokines IL-10, IL-35, and TGF-β, and the expression of the transcription factor Foxp3, which mediates Treg development, lineage commitment, and regulatory functions
[15][55]. Another marker of nTregs and pTregs is the IL-2 receptor α chain CD25
[15][55].
nTregs are positively selected in the thymus by the intermediate affinity of the TCR for self-peptides/MHC
[122][232], whereas T-cells with a high-affinity TCR antigen are eliminated and those with low-affinity differentiate into naïve T-cells
[129][239]. In humans, nTreg development seems to also depend on IL-2 and/or IL-15
[130][131][132][240,241,242].
In the thymus, a restricted number of autoreactive CD4 T-cells differentiate into nTregs, in a process called agonist selection that guarantees central tolerance to self-antigens, thus avoiding autoimmunity
[126][133][134][236,243,244]. nTregs are already in an antigen-primed or antigen-activated state in the thymus
[128][238].
pTregs differentiate from conventional CD4 T-cells in the periphery under tolerogenic conditions in secondary lymphoid tissues, in particular intestinal draining lymph nodes, upon the recognition of an antigen presented by an APC
[135][136][137][245,246,247]. pTreg differentiation requires the sustained expression of FOXP3 and is dependent on high levels of TGF-β, an absence of proinflammatory cytokines
[14][54], and the activation of naïve CD4 T-cells upon recognition of mainly exogenous antigens
[138][139][140][248,249,250]. pTreg differentiation is also facilitated by vitamin-A derived retinoic acid
[141][142][143][251,252,253].
pTregs are classified as central, effector, and tissue-resident pTregs
[144][254]. Central pTregs are considered naïve and in mice are characterized by the expression of the markers CD62Lhigh CCR7+ or CD45RAhigh CD25low. Central pTregs are the main Treg type in the circulation and in secondary lymphoid organs. The marker profile of effector Tregs, also called effector memory or activated Tregs, is CD62Llow, CCR7low, CD44hi killer cell lectin like receptor G1 (KLRG1)+, CD103+, or CD25RAlow CD25hi. Effector memory pTregs are less frequent than central pTregs and are similar to conventional activated CD4 T-cells that have had recent contact with an antigen. Tissue-resident pTregs found In non-lymphoid tissues, such as the colon, and under steady state conditions account for most pTregs in the intestine
[144][254].
Tregs are activated at much lower antigen/MHC concentrations than naïve T-cells, ensuring Treg-dependent self-tolerance
[145][255]. Tregs are a frequent immune cell population in the intestine, where they limit inflammatory CD4 T-cells
[146][147][256,257] and maintain immune homeostasis through several mechanisms
[148][258]. FoxP3+ Tregs, especially effector Tregs, are constantly proliferating under steady state conditions, likely as a consequence of identifying self-antigens and antigens derived from commensal microbes
[149][150][259,260].
The suppressor activity of Tregs is mostly mediated by cell-contact–dependent and humoral-factor–mediated mechanisms. These mechanisms include IL-2 scavenging; the secretion of regulatory cytokines, such as IL-10,
[151][261], IL-35
[152][262], and TGF-β
[153][154][263,264]; the surface expression of inhibitory molecules, such as CTLA-4 (cytotoxic lymphocyte antigen 4) and PD-1 (programmed cell death 1), TIGIT (T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains), CD39, and CD73
[155][156][265,266]; cytolysis; and metabolic control
[128][238]. Tregs also promote tissue through the release of growth factor amphiregulin
[157][267].
In the intestine, Tregs can acquire several phenotypes expressing varying levels of GATA3, Helios, and RORγt. GATA3+Helios+ Tregs seem to have a thymic origin and react to the alarmin IL-33 produced in response to tissue damage, reducing tissue injury in colitis
[158][294]. RORγt+Helios− Tregs, produced in response to intestinal microbiota, are considered pTregs and play a protective role in severe gut inflammation
[159][160][289,290]. RORγt−Helios− Tregs are more abundant in the small intestine and participate in the amelioration of allergic responses to food antigens
[161][295]. These observations indicate that Tregs are highly versatile cells that adapt to their environment in order to better contribute to tissue homeostasis. There is some interest in developing therapies to boost Treg cell number and function and thereby reduce intestinal inflammation in IBD
[144][162][163][254,296,297].
Other CD4 T-cell subsets include Foxp3- type 1 regulatory T (Tr1) cells, which secrete the suppressive cytokines IL-10 and TGF β
[19][164][26,298], and Tfh cells, which are a specialized CD4 T-cell subset involved in the induction and differentiation of B cells into plasma cells and memory cells
[165][166][167][299,300,301], cell subsets whose role in IBD has recently been reviewed
[19][144][168][169][26,254,302,303].