Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Anand Kamal Singh.
Immunotherapy has brought new hope for cancer patients. There is still a need to address major challenges including heterogeneity in response among patients, the reoccurrence of the disease, and iRAEs (immune-related adverse effects). The first critical step towards solving these issues is understanding the epigenomic events that play a significant role in the regulation of specific biomolecules in the context of the immune population present in the tumor immune microenvironment (TIME) during various treatments and responses.
- cancer
- immunotherapy
- immune checkpoint drugs
- Epigenetics
1. Introduction
Immunotherapy (a type of cancer treatment that relies on empowering the body’s own immune cells to fight cancer) has expanded to include: i—immune checkpoint/ligand inhibitors (CTLA-4, PD-1, PD-L1/L2, TIM3, and TIGIT) [1,2,3,4,5,6,7,8,9,10,11,12][1][2][3][4][5][6][7][8][9][10][11][12]; ii—adoptive T-cell transfer therapy (CAR-T, TCR-T, TIL and NK cell) [13,14,15,16,17][13][14][15][16][17]; iii—cancer vaccines (T-vec, BCG and Sipuleucel-T) [18,19][18][19]; and iv—immunomodulators (thalidomide, lenalidomide and pomalidomide) [20,21,22,23][20][21][22][23]. Immunotherapy has revolutionized cancer treatment, providing significant clinical benefits to patients with different types of cancers. However, only a small subset of patients benefit from immunotherapy, which highlights limitations of this therapy. Major limitations include the low response rate evidenced by primary/acquired resistance and iRAEs [24,25,26,27][24][25][26][27]. These limitations can be attributed to epigenetic changes acquired by the TIME that play an imperative role in the development of intra/inter tumor heterogeneity by favoring the evolution of transcriptionally distinct clonal populations of cancer cells, which ultimately aid tumor progression and development [28,29][28][29].
Epigenetic aberrations are considered hallmarks of cancer development and progression [30]. In the TIME, cancer cells escape immune-mediated cell death by utilizing epigenetic mechanisms to escape host immune recognition and immunogenicity [31,32][31][32]. In the tumor microenvironment (TME), in addition to cancer cells, immune cells also undergo various epigenetic modifications that alter their effector cytokine expression, cancer immunosurveillance, immune-checkpoint molecule expression, and tumor-associated antigen presentation with MHC molecules [33,34][33][34]. Additionally, epigenetic modulators such as DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis) can re-program the TIME to increase the susceptibility of tumor cells to cytotoxic T-cell-mediated killing, leading to enhanced anti-tumor immune responses [35,36][35][36]. Moreover, unlike genetic alterations, epigenetic modifiers can be pharmacologically altered to revert the changes acquired during cancer initiation and progression [37,38,39,40][37][38][39][40].
An improved understanding of epigenetic events related to immunotherapy resistance would be helpful in designing potential combination strategies for immunotherapy. Multiple factors including constitutive PD-L1 expression in cancer cells, a lack of tumor antigens, defective antigen presentation and processing machinery, the exhaustion of infiltrated T cells, and the presence of an immunosuppressive population—such as Tregs, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs)—could contribute to acquired resistance to immunotherapy [41,42,43][41][42][43] for TAMs [44].
2. Epigenetic Modifiers in T Cells
The functional differentiation of T cells, like short-lived effectors, long-term memory T cells, Treg, and other T-cell populations, is majorly influenced by epigenetic modifications. An increasing number of investigations support the crucial role of HATis, HDACis and HMTs in regulating the fate and function of T cells. The inhibition of HDAC1 and HDAC2 promote the differentiation of CD4+ T cells into cytotoxic CD4+ T cells [50,51][45][46]. HDAC3 is critical for the maturation of both CD4+ and CD8+T cells and the production of TNF upon TCR/CD28 stimulation [52][47]. Enrichment in the central memory and stem cell memory phenotypes of T cells is regulated by H3K4me3 modification at specific gene promoters such as TCF7, LEF1, and KLF2. Interestingly, the upregulation of H3K4me3 and the downregulation of H3K27me3 at the Gcnt1 locus were found to enhance the trafficking of memory T cells to tumor sites in an interleukin (IL)-15-dependent manner [53][48]. Scheer et al. reported that lysine methyltransferase Dot1l-dependent H3K79me2 is crucial for CD4+ T helper (Th) cell differentiation, as the loss of it was found to lead to the increased expression of Th-1-specific genes and the overproduction of IFN-γ at the expense of Th-2 cell development, advocating a central role for Dot1l in Th-2 cell lineage commitment and stability [54][49]. Another study investigated the role of menin, a major component of the trithorax group (TrxG) using Cd4-cre-driven conditional knockout (KO) mice; a deficiency in menin was shown to lead to the downregulation of Gata3 expression due to reduced levels of H3K9ac and H3K4me3 at the upstream regions of the Gata3 proximal promoter [55][50]. Interestingly, the suppression of histone H3K27 demethylases KDM6A (UTX) in mature Th-17 cells was found to reduce mitochondrial biogenesis, causing metabolic reprogramming and reducing the expression of key metabolic TFs, such as PPRC1, which ultimately showed anti-inflammatory effects [56][51]. The results of these studies reinforce the role of epigenomic events in T-cell biology.2.1. Epigenetic Modifiers in Immune Checkpoint Therapy
A critical balance between immune co-inhibitory and co-stimulatory signals in the TIME is maintained to restrict tumor development and progression (Figure 1) [57,58][52][53]. The epigenetically regulated aberrant expression of immune checkpoints (ICs), including PD-1, CTLA-4, TIM-3 (T-cell immunoglobulin and mucin-domain containing-3), LAG-3 (lymphocyte-activation gene 3), TIGIT (T-cell immunoreceptor with Ig and ITIM domains), VISTA (V-domain Ig suppressor of T-cell activation), CD276 (B7-H3), B7-H4 (VTCN1/B7x/B7S1/B7 homolog 40), IDO-1 (indoleamine 2,3-dioxygenase 1), CD161, CD38, CD93, and CD47 may result in the induction of an immune-suppressive environment, which helps tumor cells to evade immune destruction [12,59,60][12][54][55]. Targeting altered epigenetic modifications can significantly contribute to the reversal of the transcriptomic regulation of ICs and their ligands, which could help to re-establish potent host immunosurveillance mechanisms [61][56].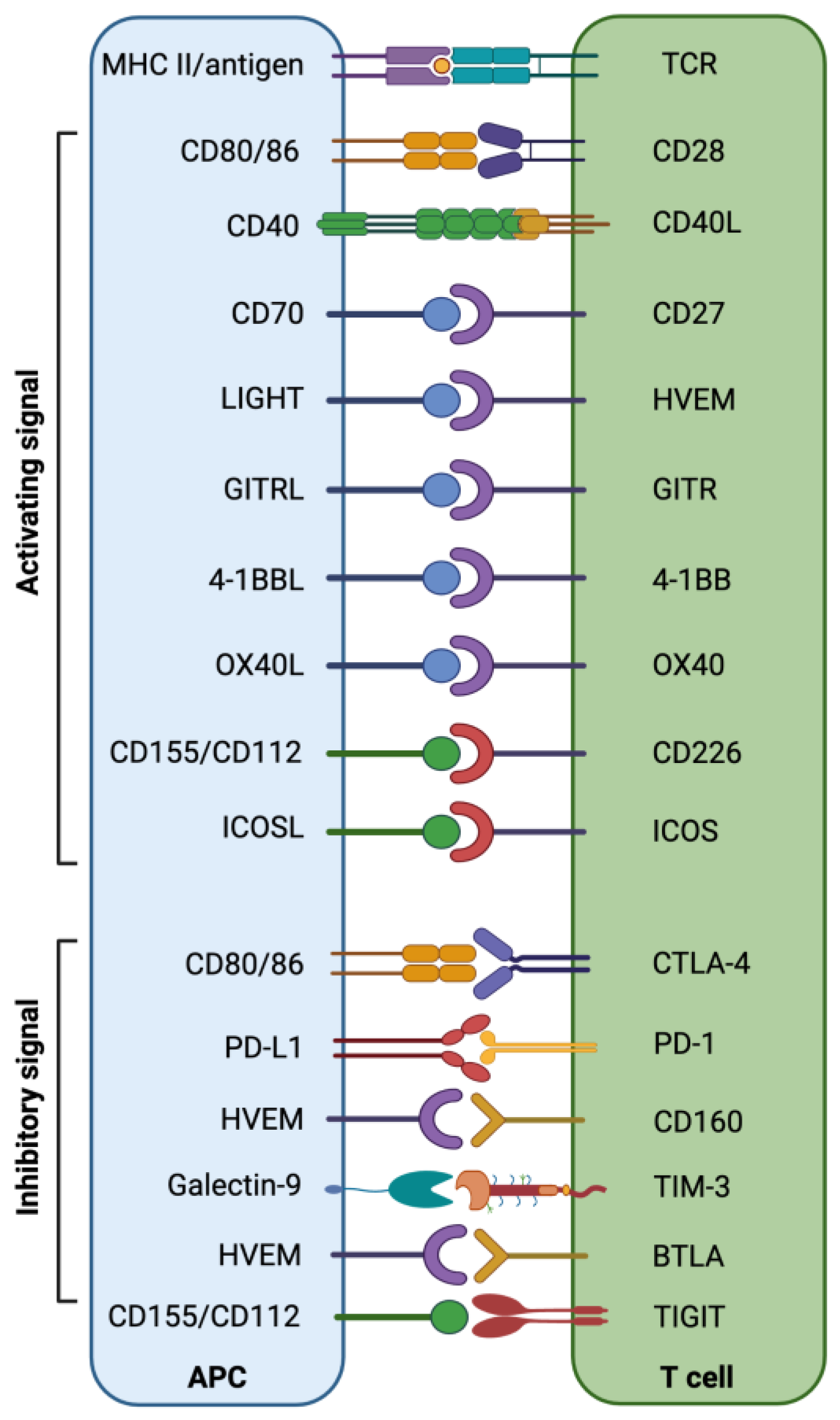
Figure 1. Interaction of co-stimulatory/inhibitory molecules between T cells and APCs/tumor cells provides an overview of the immune checkpoint/stimulatory molecules involved in the anti-tumor immune response.
2.2. Epigenetic Modifiers in Antigen Processing and Presentation
In a proper functioning immune system, T cells recognize tumor antigens based on the binding of a T-cell receptor (TCR) and a matching antigen packaged into major histocompatibility complex (MHC) proteins on APCs. Tumor cells escape immune recognition through multiple mechanisms such as alterations in antigen presentation and processing machinery (APM) or alterations in MHC class I molecules, which further impair their identification by CTLs (Figure 2).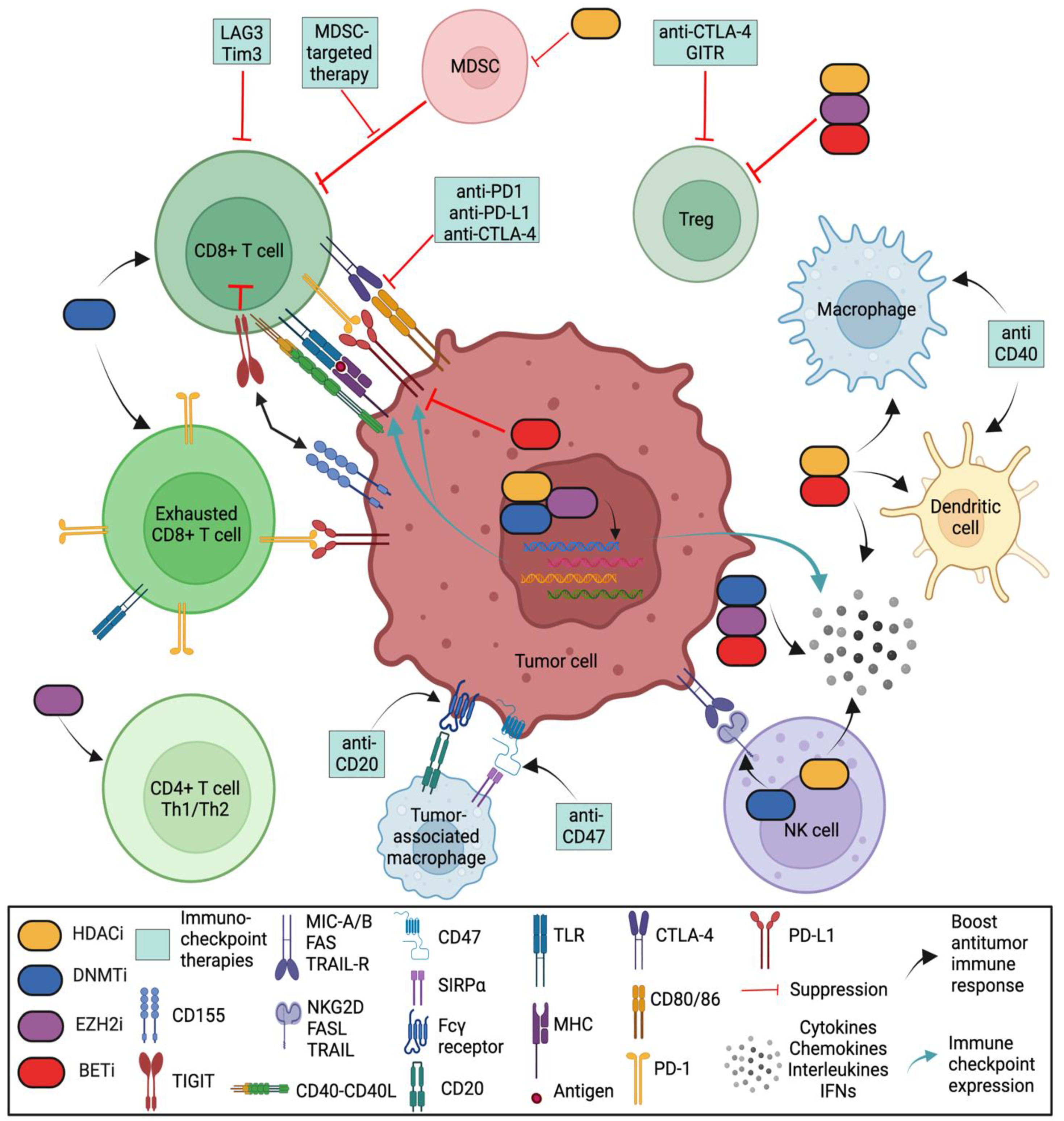
Figure 2. Role of various epigenetic modifiers in the tumor immune microenvironment. DNA methyltransferase inhibitors (DNMTis), histone deacetylase inhibitors (HDACis), an inhibitor of histone methylation on histone H3 at lysine 27 (EZH2i), and inhibitor of bromodomain and extra-terminal motif (BETi) shape the tumor-immune microenvironment by (i) increasing the number of CD8 and CD4 T cells; (ii) activating antigen processing and presentation machinery; (iii) decreasing the abundance of MDSCs and tumor-associated macrophages (TAMs); (iv) downregulating the immune checkpoint inhibitors Tim-3, Lag-3 and TIGIT; (v) upregulating immune checkpoint PD-L1 (by DNMTis, HDACis and EZH2i) and downregulating PD-L1 (by BETi); (vi) enhancing NK-mediated lysis (by HDACis) or decreasing NK cytotoxicity (by EZH2i); and (vii) upregulating inflammatory genes and pathways that control the secretion of interferons (IFNs), cytokines, and chemokines from tumor cells. (Regulatory T cells (Tregs) are a specialized subpopulation of T cells that act to suppress the immune response, thereby maintaining homeostasis and self-tolerance. It has been shown that Tregs are able to inhibit T-cell proliferation and cytokine production, as well as play a critical role in preventing autoimmunity. Tumor-associated macrophages (TAMs) are the key cells that create an immunosuppressive tumor microenvironment (TME) by producing cytokines, chemokines, and growth factors and by triggering the inhibitory immune checkpoint proteins release in T cells. Natural killer (NK) cells are effector lymphocytes of the innate immune system that control several types of tumors and microbial infections by limiting their spread and subsequent tissue damage. Cancer-associated fibroblasts (CAFs) are one of the most abundant and critical components of the tumor mesenchyme; they not only provide physical support for tumor cells but also play a key role in promoting and retarding tumorigenesis in a context-dependent manner. Recent studies have revealed their roles in immune evasion and poor responses to cancer immunotherapy).
2.3. Epigenetic Modifiers in Tumor-Infiltrating Immunosuppressive Cells
Tumor-infiltrating immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and cancer-associated fibroblasts (CAFs) inhibit T cells’ effector functionality and anti-tumor responses, which lead to the immune escape of tumors. The presence of an immunosuppressive cell population in the TIME could be a major contributory factor in ineffective ICTs [93][88]. HDACis have antitumor effects in that they reduce the number of MDSCs through various mechanisms of action such as CG-745, a class I–IIb HDACi that induces the infiltration of lymphocytes by increased antigen presentation and that decreases the amount of MDSCs by decreasing the polarization of M2 macrophages in tumors [35]. Valproic acid (VPA), a class-I HDACi, attenuates the immunosuppressive function of MDSCs by downregulating the expression of retinoblastoma 1 (Rb1), toll-like receptor 4 (TLR4), programmed cell death 1 ligand (PD-L1), and interleukin-4 receptor-alpha (IL-4Ra)/arginase [94][89]. Moreover, the combinatorial treatment of VPA and anti-PD-1 antibodies was found to repress the growth of B16F10 and EL4 tumor models by impairing tumor-infiltrating M2-MDSC accumulation in the tumor microenvironment compared with their individual therapies [95][90]. Thus, treatment with epigenetic modifiers inhibits MDSC accumulation, thereby augmenting immune checkpoint inhibitors for successful cancer treatment. Vorinostat (suberoylanilide hydroxamic acid, SAHA), a class I–II–IV HDACi, was shown to have anti-tumor potential for a 4T1 mammary mice model in which it decreased MDSC accumulation in the spleen, blood, and tumor while promoting the activation and function of CD8+ T cells [96][91]. Tregs play significant roles in inducing variety of immune responses, as determined by the expression of Foxp3, a transcription factor in natural Tregs (nTregs) in the thymus [97,98][92][93]. Extrinsic molecular signals including IL-2 and TCR, along with a network of transcription factors, are critical for regulating the expression of Foxp3 through epigenomic modulation, which ultimately determines a Treg’s phenotypic plasticity [99,100][94][95]. Epigenetic modifiers such as DNMT1 and DNMT3b are differentially bound to Foxp3 promoter and enhancer sites in nTregs compared with extrinsically induced Tregs. Importantly, DNMTis demethylate and activate the Foxp3 promoter and enhancer elements to induce Foxp3 expression and subsequently enable the induction of Foxp3-dependent, Treg-restricted sets of genes [101][96]. Demethylation in synergy with TGF-β transforms naive T cells into Tregs with high Foxp3 expression and potent, stable suppressive function [102][97]. Foxp3 expression in Treg cells was found to be significantly upregulated upon treatment with trichostatin-A (TSA), a HDACi [103][98]. Moreover, the CTLA4, PD-1, GITR and IL-10 genes are reportedly upregulated by TSA [104][99]. Ohkura et al. reported that Treg maturation, Treg-specific gene expression, and Treg-specific immunosuppressive activity involve epigenetic regulation through genome-wide CpG DNA hypomethylation pattern [105][100]. In other study, Wang et al. showed that the inhibition of EZH2, a histone-lysine N-methyltransferase enzyme, resulted in Treg-mediated pro-inflammatory activities in the TME, supporting the idea of the generation of an effector T-cell-mediated anti-tumor immune response [106][101].References
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106.
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034.
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86.
- Anderson, A.C. Tim-3, a negative regulator of anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 213–216.
- Anderson, A.C. Tim-3: An emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2014, 2, 393–398.
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009.
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57.
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937.
- Abbas, H.A.; Hao, D.; Tomczak, K.; Barrodia, P.; Im, J.S.; Reville, P.K.; Alaniz, Z.; Wang, W.; Wang, R.; Wang, F.; et al. Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat. Commun. 2021, 12, 6071.
- Wang, Y.; Zhang, H.; Liu, C.; Wang, Z.; Wu, W.; Zhang, N.; Zhang, L.; Hu, J.; Luo, P.; Zhang, J.; et al. Immune checkpoint modulators in cancer immunotherapy: Recent advances and emerging concepts. J. Hematol Oncol 2022, 15, 111.
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461.
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176.
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308.
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057.
- Qu, C.; Zhang, H.; Cao, H.; Tang, L.; Mo, H.; Liu, F.; Zhang, L.; Yi, Z.; Long, L.; Yan, L.; et al. Tumor buster—Where will the CAR-T cell therapy ‘missile’ go? Mol. Cancer 2022, 21, 201.
- Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines 2021, 9, 668.
- Kim, C.G.; Sang, Y.B.; Lee, J.H.; Chon, H.J. Combining Cancer Vaccines with Immunotherapy: Establishing a New Immunological Approach. Int. J. Mol. Sci. 2021, 22, 8035.
- Corral, L.G.; Haslett, P.A.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386.
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216.
- Haslett, P.A.; Klausner, J.D.; Makonkawkeyoon, S.; Moreira, A.; Metatratip, P.; Boyle, B.; Kunachiwa, W.; Maneekarn, N.; Vongchan, P.; Corral, L.G.; et al. Thalidomide stimulates T cell responses and interleukin 12 production in HIV-infected patients. AIDS Res. Hum. Retroviruses 1999, 15, 1169–1179.
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571.
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104.
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur J. Cancer 2016, 54, 139–148.
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168.
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chavez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suarez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38.
- Beyes, S.; Bediaga, N.G.; Zippo, A. An Epigenetic Perspective on Intra-Tumour Heterogeneity: Novel Insights and New Challenges from Multiple Fields. Cancers 2021, 13, 4969.
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727.
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978.
- Kugel, S.; Feldman, J.L.; Klein, M.A.; Silberman, D.M.; Sebastian, C.; Mermel, C.; Dobersch, S.; Clark, A.R.; Getz, G.; Denu, J.M.; et al. Identification of and Molecular Basis for SIRT6 Loss-of-Function Point Mutations in Cancer. Cell Rep. 2015, 13, 479–488.
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253.
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90.
- Callahan, S.C.; Divenko, M.; Barrodia, P.; Singh, A.K.; Arslan, E.; Liu, Z.; Yang, J.; Anvar, N.; Amit, M.; Xie, T.; et al. KMT2D Loss Promotes Head and Neck Squamous Cell Carcinoma Through Enhancer Reprogramming and Modulation of Immune Microenvironment. Biorxiv 2021.
- Kim, Y.D.; Park, S.M.; Ha, H.C.; Lee, A.R.; Won, H.; Cha, H.; Cho, S.; Cho, J.M. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J. Cancer 2020, 11, 4059–4072.
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837.
- Naik, R.R.; Singh, A.K.; Mali, A.M.; Khirade, M.F.; Bapat, S.A. A tumor deconstruction platform identifies definitive end points in the evaluation of drug responses. Oncogene 2016, 35, 727–737.
- Orouji, E.; Raman, A.T.; Singh, A.K.; Sorokin, A.; Arslan, E.; Ghosh, A.K.; Schulz, J.; Terranova, C.; Jiang, S.; Tang, M.; et al. Chromatin state dynamics confers specific therapeutic strategies in enhancer subtypes of colorectal cancer. Gut 2022, 71, 938–949.
- Singh, A.K.; Chandra, N.; Bapat, S.A. Evaluation of Epigenetic Drug Targeting of Heterogenous Tumor Cell Fractions Using Potential Biomarkers of Response in Ovarian Cancer. Clin. Cancer Res. 2015, 21, 5151–5163.
- Dong, Y.B., Jr.; Anand, K.S.; Bapat, S.; Clements, A.J. Transforming the future of treatment for ovarian cancer. Clin. Exp. Pharmacol. 2014, 4, 3.
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81.
- Pitt, J.M.; Vetizou, M.; Daillere, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723.
- Pascual-Garcia, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martinez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8(+) T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416.
- McCaw, T.R.; Goel, N.; Brooke, D.J.; Katre, A.A.; Londono, A.I.; Smith, H.J.; Randall, T.D.; Arend, R.C. Class I histone deacetylase inhibition promotes CD8 T cell activation in ovarian cancer. Cancer Med. 2021, 10, 709–717.
- Preglej, T.; Hamminger, P.; Luu, M.; Bulat, T.; Andersen, L.; Goschl, L.; Stolz, V.; Rica, R.; Sandner, L.; Waltenberger, D.; et al. Histone deacetylases 1 and 2 restrain CD4+ cytotoxic T lymphocyte differentiation. JCI Insight 2020, 5, e133393.
- Hsu, F.C.; Belmonte, P.J.; Constans, M.M.; Chen, M.W.; McWilliams, D.C.; Hiebert, S.W.; Shapiro, V.S. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 2015, 195, 1578–1590.
- Nolz, J.C.; Harty, J.T. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. J. Clin. Investig. 2014, 124, 1013–1026.
- Scheer, S.; Runting, J.; Bramhall, M.; Russ, B.; Zaini, A.; Ellemor, J.; Rodrigues, G.; Ng, J.; Zaph, C. The Methyltransferase DOT1L Controls Activation and Lineage Integrity in CD4(+) T Cells during Infection and Inflammation. Cell Rep. 2020, 33, 108505.
- Onodera, A.; Kiuchi, M.; Kokubo, K.; Kato, M.; Ogino, T.; Horiuchi, S.; Kanai, U.; Hirahara, K.; Nakayama, T. Menin Controls the Memory Th2 Cell Function by Maintaining the Epigenetic Integrity of Th2 Cells. J. Immunol. 2017, 199, 1153–1162.
- Cribbs, A.P.; Terlecki-Zaniewicz, S.; Philpott, M.; Baardman, J.; Ahern, D.; Lindow, M.; Obad, S.; Oerum, H.; Sampey, B.; Mander, P.K.; et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 6056–6066.
- O’Neill, R.E.; Cao, X. Co-stimulatory and co-inhibitory pathways in cancer immunotherapy. Adv. Cancer Res. 2019, 143, 145–194.
- Maitituoheti, M.A.S.; Tang, M.; Ho, L.-L.; Terranova, C.; Galani, K.; Keung, E.Z.; Creasy, C.A.; Wu, M.; Chen, J.; Chen, N.; et al. Enhancer Reprogramming in Melanoma Immune Checkpoint Therapy Resistance. bioRxiv 2022.
- Ni, L.; Dong, C. New B7 Family Checkpoints in Human Cancers. Mol. Cancer Ther. 2017, 16, 1203–1211.
- Wang, C.; Feng, H.; Cheng, X.; Liu, K.; Cai, D.; Zhao, R. Potential Therapeutic Targets of B7 Family in Colorectal Cancer. Front. Immunol. 2020, 11, 681.
- Saleh, R.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front. Immunol. 2020, 11, 1469.
- Buttler, C.A.; Chuong, E.B. Emerging roles for endogenous retroviruses in immune epigenetic regulation. Immunol. Rev. 2022, 305, 165–178.
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166.
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970.
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385.
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918.
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314.
- Harjes, U. SETDB1, a new target for immunotherapy. Nat. Rev. Cancer 2021, 21, 412.
- Dutta, R.; Khalil, R.; Mayilsamy, K.; Green, R.; Howell, M.; Bharadwaj, S.; Mohapatra, S.S.; Mohapatra, S. Combination Therapy of Mithramycin A and Immune Checkpoint Inhibitor for the Treatment of Colorectal Cancer in an Orthotopic Murine Model. Front. Immunol. 2021, 12, 706133.
- Andrieu, G.P.; Shafran, J.S.; Smith, C.L.; Belkina, A.C.; Casey, A.N.; Jafari, N.; Denis, G.V. BET protein targeting suppresses the PD-1/PD-L1 pathway in triple-negative breast cancer and elicits anti-tumor immune response. Cancer Lett. 2019, 465, 45–58.
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245.
- Klumper, N.; Ralser, D.J.; Bawden, E.G.; Landsberg, J.; Zarbl, R.; Kristiansen, G.; Toma, M.; Ritter, M.; Holzel, M.; Ellinger, J.; et al. LAG3 (LAG-3, CD223) DNA methylation correlates with LAG3 expression by tumor and immune cells, immune cell infiltration, and overall survival in clear cell renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000552.
- Wang, Y.; Wang, J.; Meng, J.; Jiang, H.; Zhao, J.; Qian, H.; Chen, T. Epigenetic Modification Mediates the Increase of LAG-3(+) T Cells in Chronic Osteomyelitis. Inflammation 2017, 40, 414–421.
- Li, X.; Hu, W.; Zheng, X.; Zhang, C.; Du, P.; Zheng, Z.; Yang, Y.; Wu, J.; Ji, M.; Jiang, J.; et al. Emerging immune checkpoints for cancer therapy. Acta Oncol. 2015, 54, 1706–1713.
- Moriyama, K.; Kukita, A.; Li, Y.J.; Uehara, N.; Zhang, J.Q.; Takahashi, I.; Kukita, T. Regulation of osteoclastogenesis through Tim-3: Possible involvement of the Tim-3/galectin-9 system in the modulation of inflammatory bone destruction. Lab. Invest. 2014, 94, 1200–1211.
- Zhang, H.; Song, Y.; Yang, H.; Liu, Z.; Gao, L.; Liang, X.; Ma, C. Tumor cell-intrinsic Tim-3 promotes liver cancer via NF-kappaB/IL-6/STAT3 axis. Oncogene 2018, 37, 2456–2468.
- Zhang, L.; Tian, S.; Pei, M.; Zhao, M.; Wang, L.; Jiang, Y.; Yang, T.; Zhao, J.; Song, L.; Yang, X. Crosstalk between histone modification and DNA methylation orchestrates the epigenetic regulation of the costimulatory factors, Tim3 and galectin9, in cervical cancer. Oncol. Rep. 2019, 42, 2655–2669.
- Zhang, L.; Tian, S.; Zhao, M.; Yang, T.; Quan, S.; Yang, Q.; Song, L.; Yang, X. SUV39H1-DNMT3A-mediated epigenetic regulation of Tim-3 and galectin-9 in the cervical cancer. Cancer Cell Int. 2020, 20, 325.
- Niebel, D.; Frohlich, A.; Zarbl, R.; Fietz, S.; de Vos, L.; Vogt, T.J.; Dietrich, J.; Sirokay, J.; Kuster, P.; Saavedra, G.; et al. DNA methylation regulates TIGIT expression within the melanoma microenvironment, is prognostic for overall survival, and predicts progression-free survival in patients treated with anti-PD-1 immunotherapy. Clin. Epigenetics 2022, 14, 50.
- Gibbs, Z.A.; Whitehurst, A.W. Emerging Contributions of Cancer/Testis Antigens to Neoplastic Behaviors. Trends Cancer 2018, 4, 701–712.
- Wang, C.; Gu, Y.; Zhang, K.; Xie, K.; Zhu, M.; Dai, N.; Jiang, Y.; Guo, X.; Liu, M.; Dai, J.; et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat. Commun. 2016, 7, 10499.
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.W.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. 2018, 24, 1019–1029.
- Ishihara, M.; Kitano, S.; Kageyama, S.; Miyahara, Y.; Yamamoto, N.; Kato, H.; Mishima, H.; Hattori, H.; Funakoshi, T.; Kojima, T.; et al. NY-ESO-1-specific redirected T cells with endogenous TCR knockdown mediate tumor response and cytokine release syndrome. J. Immunother. Cancer 2022, 10, e003811.
- Grunewald, C.M.; Schulz, W.A.; Skowron, M.A.; Hoffmann, M.J.; Niegisch, G. Tumor immunotherapy—The potential of epigenetic drugs to overcome resistance. Transl. Cancer Res. 2018, 7, 1151–1160.
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654.
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 2000, 165, 7017–7024.
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607.
- Maeda, T.; Towatari, M.; Kosugi, H.; Saito, H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood 2000, 96, 3847–3856.
- Wang, L.X.; Mei, Z.Y.; Zhou, J.H.; Yao, Y.S.; Li, Y.H.; Xu, Y.H.; Li, J.X.; Gao, X.N.; Zhou, M.H.; Jiang, M.M.; et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS ONE 2013, 8, e62924.
- Federico, A.; Steinfass, T.; Larribere, L.; Novak, D.; Moris, F.; Nunez, L.E.; Umansky, V.; Utikal, J. Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Malignant Melanoma. Mol. Ther. Oncolytics 2020, 18, 83–99.
- Lin, J.; Guo, D.; Liu, H.; Zhou, W.; Wang, C.; Muller, I.; Kossenkov, A.V.; Drapkin, R.; Bitler, B.G.; Helin, K.; et al. The SETDB1-TRIM28 Complex Suppresses Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 1413–1424.
- Truong, A.S.; Zhou, M.; Krishnan, B.; Utsumi, T.; Manocha, U.; Stewart, K.G.; Beck, W.; Rose, T.L.; Milowsky, M.I.; He, X.; et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J. Clin. Invest. 2021, 131, e138560.
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550.
- Xie, Z.; Ago, Y.; Okada, N.; Tachibana, M. Valproic acid attenuates immunosuppressive function of myeloid-derived suppressor cells. J. Pharmacol. Sci. 2018, 137, 359–365.
- Xie, Z.; Ikegami, T.; Ago, Y.; Okada, N.; Tachibana, M. Valproic acid attenuates CCR2-dependent tumor infiltration of monocytic myeloid-derived suppressor cells, limiting tumor progression. Oncoimmunology 2020, 9, 1734268.
- Wang, H.F.; Ning, F.; Liu, Z.C.; Wu, L.; Li, Z.Q.; Qi, Y.F.; Zhang, G.; Wang, H.S.; Cai, S.H.; Du, J. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. 2017, 66, 355–366.
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22, 329–341.
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 2005, 201, 1061–1067.
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290.
- Kim, H.P.; Leonard, W.J. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: A role for DNA methylation. J. Exp. Med. 2007, 204, 1543–1551.
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009, 182, 259–273.
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886.
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307.
- Zhang, H.; Xiao, Y.; Zhu, Z.; Li, B.; Greene, M.I. Immune regulation by histone deacetylases: A focus on the alteration of FOXP3 activity. Immunol. Cell Biol. 2012, 90, 95–100.
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799.
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274.
More