Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Tamanna Roshan Lal.
Asthma is a heterogenous disorder driven by inflammatory mechanisms that result in multiple phenotypes. Given the complex nature of this condition, metabolomics is being used to delineate the pathobiology of asthma. Metabolomics is the study of metabolites in biology, which includes biofluids, cells, and tissues. These metabolites have a vital role in a disease as they contribute to the pathogenesis of said condition.
- asthma
- metabolomic
- macrometabolic
1. Introduction
Traditionally, asthma was thought to be a single entity described as bronchial hyperactivity with atopy and characterized by high peripheral and airway eosinophilic inflammation [1,2,3][1][2][3]. However, over the past two decades, there is increased awareness that asthma is a heterogenous disorder. It includes several distinct subtypes, including varying phenotypes (e.g., young atopic, obese middle-aged, and elderly), some of which can be distinguished by differences in biomarkers (i.e., endotypes) [2]. With the continued increase in childhood obesity, one phenotype that is garnering a lot of interest is the “obese asthma” phenotype [4,5][4][5]. Studies show that obesity is a risk factor for asthma, which is more severe than healthy-weight asthma, with higher need for hospitalization in the intensive care unit, greater pulmonary function deficits, and decreased responsiveness to existent management options [6,7][6][7]. However, the distinguishing features and the biomarkers for disease is not well understood.
The existing literature highlights the vital role of metabolomics. It is an important emerging method of understanding phenotypes of complex disorders such as obesity-related asthma by distinguishing metabolic patterns to facilitate a better understanding of disease phenotype and, thereby, the development of the personalized management of asthma. The emerging studies of metabolomics in asthma are defining various phenotypes of this complex disorder as well as delineating the pathogenesis of disease from a metabolic perspective [8]. The current literature suggests that metabolic derangements are routinely present in patients with the “obese asthma” phenotype. The metabolic derangements overlap with those present in metabolic syndrome, which includes five categories: abdominal obesity, hypertriglyceridemia, reduced high-density lipoprotein (HDL) levels, hypertension, and insulin resistance/hyperglycemia [9]. While people continue to further understand the etiology of metabolic syndrome, there are data that show it is associated with worsening lung function [10] by modulating airway responses in asthma [11]. For example, neuropeptide glucagon-like peptide-1 (GLP-1) and nitric oxide (NO) signaling pathways are shown to be dysregulated in metabolic syndrome [12].
2. Macrometabolic Associations in Asthma
2.1. Altered Carbohydrate Metabolism Is Associated with Disease Burden in Obesity-Related Asthma
Metabolic dysregulation, including insulin resistance (IR) and altered glucose metabolism, have been consistently associated with childhood and adult asthma. IR is a well-described obesity-mediated metabolic complication that has been linked with obesity-related asthma [13,14,15][13][14][15]. There is a correlation between obesity-mediated inflammation and IR, which likely contributes to asthma in obese children [16,17][16][17]. Children with asthma are prone to have higher prevalence of IR compared to patients without asthma [18,19,20][18][19][20] as quantified by acanthosis nigricans [18,19,20][18][19][20]. There is also evidence of an inverse relationship between IR and pulmonary function [11]. Higher levels of IR are associated with a lower FEV1/FVC ratio, the ratio of the forced expiratory volume in the first one second to the forced vital capacity of the lungs, which is the most consistently used pulmonary function index for the quantification of airflow obstruction (Figure 1).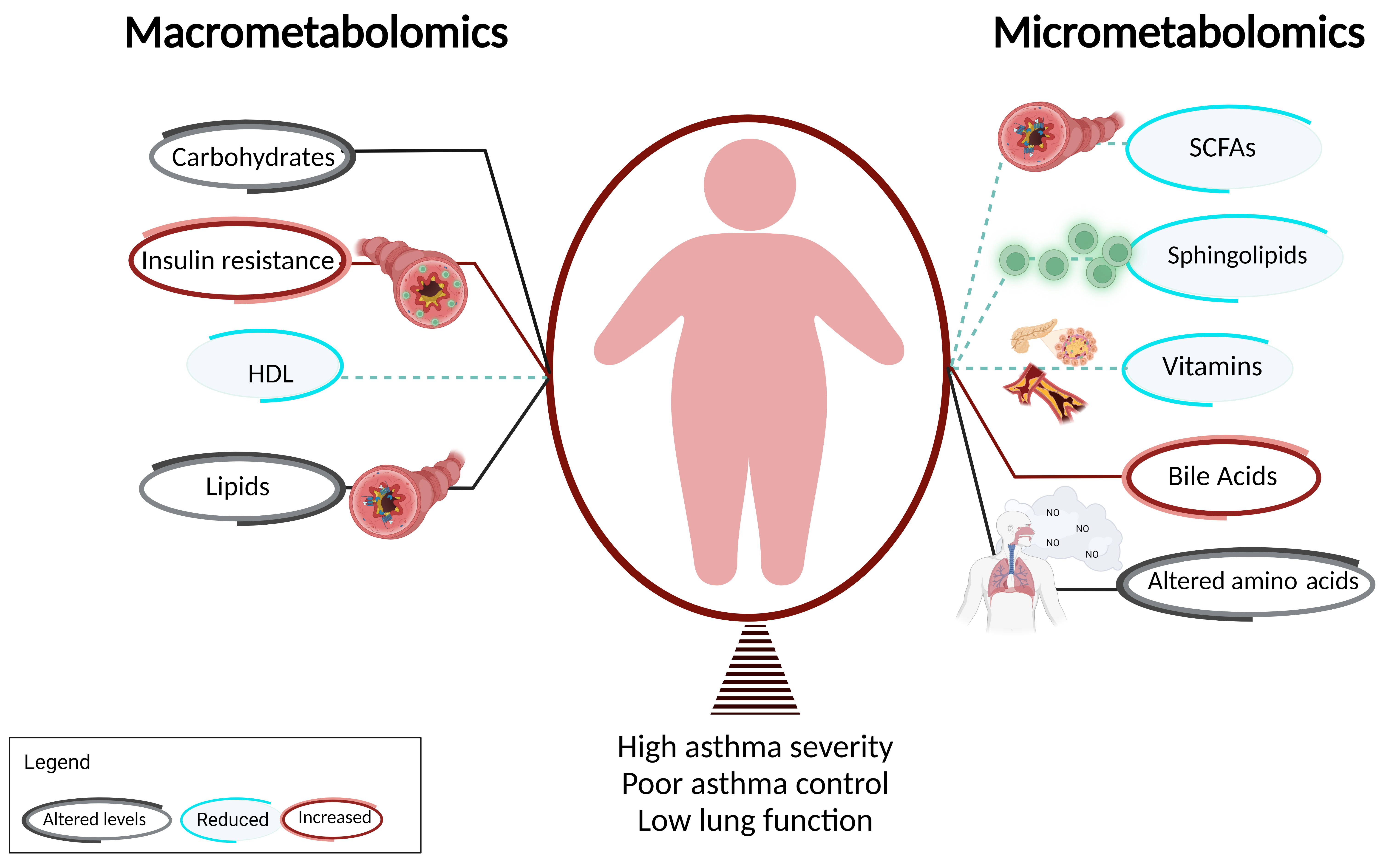
Figure 1. Macrometabolites, micrometabolites, and obesity-related asthma. This figure summarizes the association of obesity-related asthma with macrometabolites and micrometabolites and their underlying mechanisms. T cells are shown in green and FFA receptors are marked in blue.
2.2. Insulin Resistance Influences Asthma Phenotype Partly via Effects on Airway Smooth Muscle
The pathophysiologic mechanisms by which IR impacts pulmonary physiology have been investigated to some degree. Airway smooth muscle (ASM) cells express insulin receptors and develop a pro-contractile phenotype when exposed to insulin; this effect may be more pronounced in obesity [26,27][26][27]. Pharmacological agents that alter insulin and glucose metabolism have been linked with decreased disease burden in obese asthma [28,29,30][28][29][30]. Since obesity-induced IR is associated with compensatory hyperinsulinemia, it can be speculated that, like muscle, the ASM retains insulin sensitivity and may, therefore, have an augmented response to insulin exposure. However, the direct impact of IR and hyperinsulinemia on lung tissue is poorly understood. There are few studies on human tissue, but the mechanisms by which IR and hyperinsulinemia affect lung tissue have been investigated to some extent in murine models. A 16-h exposure to insulin in both obese-prone and obese-resistant murine models potentiated bronchoconstriction with ASM contraction induced by vagal stimulation, which was blocked by atropine, supporting a role of muscarinic receptors [31]. The pro-contractile effect occurred as early as 30 min after insulin exposure. This effect was seen in rat tracheal smooth muscle, which was eventually replicated in human ASM [31]. Validating these findings, Nie et al. showed that AHR to vagal stimulation increased in obese-prone animals on a high fat diet (HFD) compared to those fed a low-fat diet or obese-resistant rats; these effects were independent of body weight and body fat [32]. Suppressing insulin significantly reduced vagal-induced bronchoconstriction in rats on HFD, suggesting that hyperinsulinemia, rather than obesity alone, underlie obesity-induced bronchoconstriction via parasympathetic nerves. Similar findings have been reported with bovine ASM [33,34][33][34]. Further, since AHR is a balance between muscarinic and adrenergic receptor activation, Xu et al. investigated insulin-mediated effects on β2 adrenergic receptors (β2AR) and found a novel role for transactivation of a G protein-coupled receptor kinase 2 (GRK2)-dependent β2AR-Gi-ERK1/2 cascade in ASM cells that was associated with reduced cAMP accumulation with impaired ASM relaxation [35]. Together, these studies demonstrate that effects of insulin on AHR are mediated by dysfunction of both the M2 muscarinic receptor and the β2AR. Highlighting species-specific differences in ASM responsiveness to insulin, a single study that investigated ASM responses from obese individuals found that a 24-h insulin treatment of ASM was not associated with increased calcium release in response to carbachol [27]. While differences in the duration and dose of insulin exposure may potentially explain these differences, these incongruent results identify a need for further investigation, specifically on human ASM. In addition to AHR, hyperinsulinemia has been associated with increased α-smooth muscle actin and β-catenin in lung tissue, suggesting proliferation of ASM cells. These effects decreased in response to PI3K-inhibition [36]. These observations were validated in human ASM cells, wherein increasing concentrations of insulin for 72 h was associated with an eightfold increase in cells, and deposition of collagen, which is involved in airway remodeling [36]. This study was the first to define potentially irreversible pro-constrictive and profibrotic effects of prolonged insulin exposure on ASM mediated by PI3/Akt pathway activation. In keeping with these effects of insulin on ASM, treatment with anti-diabetic medications, including metformin, a biguanide, and albiglutide, a glucagon-like peptide-1 receptor (GLP-1R) agonist, have been associated with decreased asthma disease burden [28,29][28][29]. However, pioglitazone, a thiazolidinedione, also used as an anti-diabetic medication, does not decrease asthma burden [37]. These discrepant results may be due to differences in mechanisms of action of these anti-diabetic medications. For instance, murine studies on the effects of insulin have reported the altered activation of molecules such as the insulin receptor substrate (IRS-1) and AKT downstream of insulin through altered glycation and nitration, which is due to enhanced oxidative stress [38]. Based on this, glycation modulation by GLP-1R agonists [39] and decrease in oxidative stress by metformin [40], may explain their association with improved asthma control in individuals with obesity and asthma [30]. Metformin also decreases airway inflammation and airway remodeling by modulating 5′-adenosine monophosphate-activated protein kinase α (AMPK-α) activity [41,42][41][42]. Pioglitazone, a thiazolidinedione, influences insulin sensitivity via modified fatty acid metabolism. Although metformin is a standard therapy for prediabetes and GLP-1R agonist use is increasing in children [43[43][44],44], their effectiveness in childhood obesity-related asthma is not known. Given their effectiveness in decreasing asthma burden in adults and known safety profile in children, it can be proposed that these medication classes may serve as potential novel therapy for childhood obesity-related asthma.2.3. Dyslipidemia and Dysregulation of Fatty Acids Are Associated with Obesity-Related Asthma
Children with asthma are prone to have higher prevalence of dyslipidemia compared to patients without asthma [18,19,20][18][19][20] as quantified by decreased high-density lipoprotein (HDL) in the context of increased low-density lipoprotein (LDL), total cholesterol levels, and triglycerides [18,19,20][18][19][20] (Figure 1). There is also evidence of an inverse relationship between dyslipidemia with pulmonary function [11]. Lower levels of HDL are associated with lower FEV1/FVC ratio. Serum HDL is negatively associated with the Th1/Th2 ratio and patrolling monocytes, which are elevated in obesity [13]. In addition to lipoproteins, free fatty acids (FFA), classified based on the length of their carbon chains as small-chain, medium-chain, or long-chain, influence the pathogenesis of metabolic diseases, including obesity, type II diabetes, and atherosclerosis, and have been linked with respiratory diseases, including asthma. Medium-chain FFA (MCFAs) and long-chain FFA (LCFAs) are derived through de novo synthesis or fat intake [45], while small-chain FFA (SCFAs) are synthesized by gut microbiota through fermentation of undigested carbohydrates in the cecum and colon [46]. LCFAs are chronically elevated in obese individuals due to increased adipose tissue [47]. Several G-protein-coupled receptors (GPCRs) function as specific receptors for FFAs (FFARs) such that LCFAs and MCFAs function as ligands for FFAR1 (GPR40) and FFAR4 (GPR120), while SCFAs act as ligands for FFAR2 (GPR43) and FFAR3 (GP141). FFAR expression have been found on airway structural cells including ASM cells. While FFAR1 has been investigated, less is known about the effects of FFAR2, 3, and 4 in response to FFAs. Therefore, there is a need to define the contribution of FFA and FFARs in the pathogenesis of obesity-related asthma.2.4. Dyslipidemia Influences Asthma Phenotype Partly via Its Effects on FFA Receptors
FFAR1 protein is expressed in human ASM cells and bronchial epithelial cells [47], with limited investigation of their role in human ASM function. Activation of FFAR1 by LCFAs or FFAR1/4 agonist (GW9508), caused a sustained increase in acetylcholine-induced contractile tone with a rapid and transient rise in intracellular calcium in human ASM cells in a dose-dependent manner. This was suppressed in FFAR1-knockdown cells compared to the control [47]. While LCFAs additionally attenuated the relaxant effect of isoproterenol, a β2AR agonist, GW9508 did not exert any effect of isoproterenol-induced relaxation, suggesting differences potentially due to the involvement of FFAR1 with or without FFAR4. In addition to increased intracellular calcium, LCFAs and GW9508 induced actin reorganization, which is required for smooth muscle contraction. Collectively, the results suggest that LCFAs activate FFAR1 to induce calcium mobilization in human ASM cells via the Gq-PLC/IP3 pathway. Investigating the effects of FFRA1 activation on ASM proliferation in human and murine models, Matoba et al. additionally reported that LCFA and GW9508 exposure for 48 h was associated with ERK and Akt phosphorylation in human ASM cells, with downstream mTORC1 activation, which induced ASM proliferation [48]. This effect of FFAR1 activation induced ASM proliferation was inhibited by pre-treatment with Gi protein inhibitor (PTX), which abolished both ERK and Akt phosphorylation. Contrasting results were reported by Xu et al., where pre-treatment with TAK875 (another selective FFAR1 agonist) attenuated histamine-induced myosin light chain (MLC) phosphorylation and carbachol-induced MLC phosphorylation, and inhibited ASM shortening in β2AR-desensitized human ASM cells, independent of cAMP levels and PI3/Akt activation [49]. Considering the limited literature, these studies suggest that the differences possibly stem from differences between donors and differences in the FFAR1 agonists, as well as the role of myosin light chain phosphorylation in responsiveness to FFAR receptor engagement by different ligands.2.5. Therapies for Dyslipidemia/FFAs Are Effective in Decreasing Disease Burden of Obesity-Related Asthma
The role of FFAs in causing AHR by modulation of ASM cells, and the effect of statins in lowering FFAs [50], support epidemiological reports of the beneficial effect of statins in the context of asthma [51]. A meta-analysis of randomized controlled trials (RCTs) demonstrated that statins improve Asthma Control Test (ACT) scores and were independently associated with reduction in asthma-related ED visits and hospitalizations [51]. However, the meta-analysis did not find an association of statin use with pre- and post-bronchodilator FEV1, and peak expiratory flow (PEF), suggesting that the effect of statins on FFAs may not directly translate into a decrease in AHR. Given the dearth of mechanistic studies of how lipid lowering agents influence asthma disease burden, it can be speculated that mechanistic differences between lipid lowering agents may underlie their effectiveness in decreasing disease burden without influencing pulmonary function. These hypotheses need to be investigated to elucidate mechanistic effects of lipid lowering medications, which will inform their choice for the management of obesity-related asthma. The recent discovery of anti-inflammatory and immunomodulatory properties of statins beyond their cholesterol-lowering function, has resulted in a novel and innovative research avenue relevant to lung diseases. Experimental mouse models have demonstrated that simvastatin attenuates eosinophilic airway inflammation via inhibition of HMG-CoA; however, AHR and lung compliance variations were mevalonate- and HMG-CoA-independent [52], suggesting that statins may not directly influence AHR, but may influence it via their immune modulation. Similar additional mechanistic studies that explain how lipid lowering agents influence asthma disease burden are needed before they can be considered for therapy for childhood obesity-related asthma.References
- Cleave, J.V.; Gortmaker, S.L.; Perrin, J.M. Dynamics of obesity and chronic health conditions among children and youth. JAMA 2010, 303, 623–630.
- Kuruvilla, M.E.; Lee, F.E.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Al-lergy Immunol. 2019, 56, 219–233.
- Bousquet, J.; Chanez, P.; Lacoste, J.Y.; Barnéon, G.; Ghavanian, N.; Enander, I.; Venge, P.; Ahlstedt, S.; Simony-Lafontaine, J.; Godard, P.; et al. Eosinophilic inflammation in asthma. N. Engl. J. Med. 1990, 323, 1033–1039.
- Ray, A.; Oriss, T.B.; Wenzel, S.E. Emerging molecular phenotypes of asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L130–L140.
- Sutherland, E.R.; Goleva, E.; King, T.S.; Lehman, E.; Stevens, A.D.; Jackson, L.P.; Stream, A.R.; Fahy, J.V. Cluster Analysis of Obesity and Asthma Phenotypes. PLoS ONE 2012, 7, e36631.
- Stanley, A.H.; Demissie, K.; Rhoads, G.G. Asthma Development with Obesity Exposure: Observations from the Cohort of the National Health and Nutrition Evaluation Survey Epidemiologic Follow-up Study (NHEFS). J. Asthma 2005, 42, 97–99.
- Chen, Y.; Dales, R.; Jiang, Y. The Association Between Obesity and Asthma Is Stronger in Nonallergic Than Allergic Adults. Chest 2006, 130, 890–895.
- Wang, C.; Jiang, S.; Zhang, S.; Ouyang, Z.; Wang, G.; Wang, F. Research Progress of Metabolomics in Asthma. Metabolites 2021, 11, 567.
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.-C.; James, W.P.T.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645.
- Forno, E.; Han, Y.Y.; Muzumdar, R.H.; Celedón, J.C. Insulin resistance, metabolic syndrome, and lung function in US adolescents with and without asthma. J. Allergy Clin. Immunol. 2015, 136, 304–311.e308.
- Kim, M.; Choi, S.; Choi, S.-H.; Shin, S.-H.; Kim, S.K.; Shim, Y.S.; Jeon, Y.H. Metabolic syndrome and lung function in Korean children and adolescents: A cross-sectional study. Sci. Rep. 2019, 9, 15646.
- McCravy, M.; Ingram, J.L.; Que, L.G. Dysregulated Metabolism in the Pathophysiology of Non-Allergic Obese Asthma. J. Asthma Allergy 2021, 14, 179–186.
- Rastogi, D.; Fraser, S.; Oh, J.; Huber, A.M.; Schulman, Y.; Bhagtani, R.H.; Khan, Z.S.; Tesfa, L.; Hall, C.B.; Macian, F. Inflammation, metabolic dysregulation, and pulmonary function among obese urban ado-lescents with asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 149–160.
- Cardet, J.C.; Ash, S.; Kusa, T.; Camargo, C.A.; Jr Israel, E. Insulin resistance modifies the association between obesity and current asthma in adults. Eur. Respir. J. 2016, 48, 403–410.
- Forno, E. Asthma and diabetes: Does treatment with metformin improve asthma? Respirology 2016, 21, 1144–1145.
- Nathan, B.M.; Moran, A. Metabolic complications of obesity in childhood and adolescence: More than just diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 21–29.
- Gutierrez, D.A.; Puglisi, M.J.; Hasty, A.H. Impact of increased adipose tissue mass on inflammation, insulin resistance, and dyslipidemia. Curr. Diab. Rep. 2009, 9, 26–32.
- Al-Shawwa, B.A.; Al-Huniti, N.H.; DeMattia, L.; Gershan, W. Asthma and insulin resistance in morbidly obese children and adolescents. J. Asthma 2007, 44, 469–473.
- Cottrell, L.; Neal, W.A.; Ice, C.; Perez, M.K.; Piedimonte, G. Metabolic abnormalities in children with asthma. Am. J. Respir. Crit. Care Med. 2011, 183, 441–448.
- Arshi, M.; Cardinal, J.; Hill, R.J.; Davies, P.S.; Wainwright, C. Asthma and insulin resistance in children. Respirology 2010, 15, 779–784.
- Forno, E. A Potential New Treatment Option for Asthma in the Setting of Obesity or Insulin Resistance? Am. J. Respir. Crit. Care Med. 2021, 203, 788–789.
- Kim, S.H.; Kim, H.S.; Min, H.K.; Lee, S.W. Association between insulin resistance and lung function trajectory over 4 years in South Korea: Community-based prospective cohort. BMC Pulm. Med. 2021, 21, 110.
- McMahon, G.T.; Arky, R.A. Inhaled insulin for diabetes mellitus. N. Engl. J. Med. 2007, 356, 497–502.
- Rastogi, D.; Bhalani, K.; Hall, C.B.; Isasi, C.R. Association of pulmonary function with adiposity and metabolic abnormalities in urban minority adolescents. Ann. Amer. Thor. Soc. 2014, 11, 744–752.
- Karampatakis, N.; Karampatakis, T.; Galli-Tsinopoulou, A.; Kotanidou, E.P.; Tsergouli, K.; Eboriadou-Petikopoulou, M.; Haidopoulou, K. Impaired glucose metabolism and bronchial hyperrespon-siveness in obese prepubertal asthmatic children. Pediatr. Pulmonol. 2017, 52, 160–166.
- Schaafsma, D.; Gosens, R.; Ris, J.M.; Zaagsma, J.; Meurs, H.; Nelemans, S.A. Insulin induces airway smooth muscle contraction. Br. J. Pharmacol. 2007, 150, 136–142.
- Orfanos, S.; Jude, J.; Deeney, B.T.; Cao, G.; Rastogi, D.; Van Zee, M.; Pushkarsky, I.; Munoz, H.E.; Damoiseaux, R.; Di Carlo, D.; et al. Obesity increases airway smooth muscle responses to contractile agonists. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L673–L681.
- Wu, T.D.; Keet, C.A.; Fawzy, A.; Segal, J.B.; Brigham, E.P.; McCormack, M.C. Association of Metformin Initiation and Risk of Asthma Exacerbation. A Claims-based Cohort Study. Ann. Am. Thorac. Soc. 2019, 16, 1527–1533.
- Foer, D.; Beeler, P.E.; Cui, J.; Karlson, E.W.; Bates, D.W.; Cahill, K.N. Asthma Exacerbations in Patients with Type 2 Diabetes and Asthma on Glucagon-like Peptide-1 Receptor Agonists. Am. J. Respir. Crit. Care Med. 2021, 203, 831–840.
- Rastogi, D. Evidence Builds for a Role of Metformin in Asthma Management. Ann. Am. Thorac. Soc. 2019, 16, 1497–1499.
- Proskocil, B.J.; Calco, G.N.; Nie, Z. Insulin acutely increases agonist-induced airway smooth muscle contraction in humans and rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L545–L556.
- Nie, Z.; Jacoby, D.B.; Fryer, A.D. Hyperinsulinemia potentiates airway responsiveness to parasympathetic nerve stimulation in obese rats. Am. J. Respir. Cell Mol. Biol. 2014, 51, 251–261.
- Schaafsma, D.; McNeill, K.D.; Stelmack, G.L.; Gosens, R.; Baarsma, H.; Dekkers, B.G.J.; Frohwerk, E.; Penninks, J.-M.; Sharma, P.; Ens, K.M.; et al. Insulin increases the expression of contractile phenotypic markers in airway smooth muscle. Am. J. Physiol. Cell Physiol. 2007, 293, C429–C439.
- Gosens, R.; Nelemans, S.A.; Hiemstra, M.; Grootte Bromhaar, M.M.; Meurs, H.; Zaagsma, J. Insulin induces a hypercontractile airway smooth muscle phenotype. Eur. J. Pharmacol. 2003, 481, 125–131.
- Xu, R.; Gopireddy, R.R.; Wu, Y.; Wu, L.; Tao, X.; Shao, J.; Wang, W.; Li, L.; Jovanovic, A.; Xu, B.; et al. Hyperinsulinemia promotes heterologous desensitization of beta2 adrenergic receptor in airway smooth muscle in obesity. FASEB J. 2020, 34, 3996–4008.
- Singh, S.; Bodas, M.; Bhatraju, N.K.; Pattnaik, B.; Gheware, A.; Parameswaran, P.K.; Thompson, M.; Freeman, M.; Mabalirajan, U.; Gosens, R.; et al. Hyperinsulinemia adversely affects lung structure and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L837–L845.
- Dixon, A.E.; Subramanian, M.; DeSarno, M.; Black, K.; Lane, L.; Holguin, F. A pilot randomized controlled trial of pioglitazone for the treatment of poorly controlled asthma in obesity. Respir. Res. 2015, 16, 143.
- Andre, D.M.; Calixto, M.C.; Sollon, C.; Alexandre, E.C.; Tavares, E.B.G.; Naime, A.C.A.; Anhê, G.F.; Antunes, E. High-fat diet-induced obesity impairs insulin signaling in lungs of aller-gen-challenged mice: Improvement by resveratrol. Sci. Rep. 2017, 7, 17296.
- Nguyen, D.V.; Linderholm, A.; Haczku, A.; Kenyon, N. Glucagon-like peptide 1: A potential anti-inflammatory pathway in obesity-related asthma. Pharmacol. Ther. 2017, 180, 139–143.
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell. Endocrinol. 2020, 500, 110628.
- Park, C.S.; Bang, B.-R.; Kwon, H.-S.; Moon, K.-A.; Kim, T.-B.; Lee, K.-Y.; Moon, H.-B.; Cho, Y.S. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem. Pharmacol. 2012, 84, 1660–1670.
- Ma, W.; Jin, Q.; Guo, H.; Han, X.; Xu, L.; Lu, S.; Wu, C. Metformin Ameliorates Inflammation and Airway Remodeling of Experimental Allergic Asthma in Mice by Restoring AMPKalpha Activity. Front. Pharmacol. 2022, 13, 780148.
- Esquivel Zuniga, R.; DeBoer, M.D. Prediabetes in Adolescents: Prevalence, Management and Diabetes Prevention Strategies. Diabetes Metab. Syndr. Obes. 2021, 14, 4609–4619.
- Bensignor, M.O.; Wolf, J.M.; Rudser, K.D.; Kelly, A.S.; Arslanian, S. Glucagon-like peptide-1 receptor agonist prescribing patterns in adolescents with type 2 diabetes. Diabetes Obes. Metab. 2022, 24, 1380–1384.
- Mizuta, K.; Matoba, A.; Shibata, S.; Masaki, E.; Emala, C.W., Sr. Obesity-induced asthma: Role of free fatty acid receptors. Jpn. Dent. Sci. Rev. 2019, 55, 103–107.
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210.
- Mizuta, K.; Zhang, Y.; Mizuta, F.; Hoshijima, H.; Shiga, T.; Masaki, E.; Charles, W.E., Sr. Novel identification of the free fatty acid receptor FFAR1 that promotes contraction in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L970–L982.
- Matoba, A.; Matsuyama, N.; Shibata, S.; Masaki, E.; Emala, C.W.; Mizuta, K., Sr. The free fatty acid receptor 1 promotes airway smooth muscle cell proliferation through MEK/ERK and PI3K/Akt signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L333–L348.
- Xu, S.; Schwab, A.; Karmacharya, N.; Cao, G.; Woo, J.; Kim, N.; An, S.S.; Panettieri, R.A., Jr.; Jude, J.A. FFAR1 activation attenuates histamine-induced myosin light chain phosphorylation and cortical tension development in human airway smooth muscle cells. Respir. Res. 2020, 21, 317.
- Worgall, T.S. Sphingolipids and Asthma. Adv. Exp. Med. Biol. 2022, 1372, 145–155.
- Perzanowski, M.S.; Ono, J.G.; Acosta, L.M.; Kim, B.I.; Divjan, A.; Miller, R.; Rundle, A.; Worgall, S.; Worgall, T.S. Distinct Serum Sphin-golipid Profiles among School-aged Children with Exercise-induced Wheeze and Asthma Persistence. Am. J. Respir. Crit. Care Med. 2017, 195, 1068–1070.
- Rago, D.; Pedersen, C.T.; Huang, M.; Kelly, R.S.; Gürdeniz, G.; Brustad, N.; Knihtilä, H.; Lee-Sarwar, K.A.; Morin, A.; Rasmussen, M.A.; et al. Characteristics and Mechanisms of a Sphingolipid-associated Childhood Asthma Endotype. Am. J. Respir. Crit. Care Med. 2021, 203, 853–863.
More