Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Nathalia Soares da Cruz.
Liver cancer is one of the most lethal malignancies and is commonly diagnosed as hepatocellular carcinoma (HCC), a tumor type that affects about 90% of patients. Non-alcoholic steatohepatitis (NASH) and obesity are both risk factors for this disease. HCC initiation and progression are deeply linked with changes in the hepatic microenvironment, with cytokines playing key roles. The understanding of the pathogenic pathways that connect these disorders to liver cancer remains poor.
- HCC
- NASH
- obesity
- inflammation
- inflammasomes
1. Introduction
According to data from the Global Burden of Disease (GBD), in 2019 liver cancer was the eighth death-related type of cancer in the world. Hepatocellular carcinoma (HCC) is the most common primary liver cancer and covers about 80–90% of cases [1]. This cancer is aggressive and impacts liver hepatocytes. Its diagnosis can be made through magnetic resonance imaging, ultrasound, and serological tests that identify biomarkers such as alpha-fetoprotein (AFP), des-gamma-carboxy-prothrombin (DCP) [2], and, more recently, micro RNAs, such as miR-25 [3]. Biopsy is another option, but it is avoided due to its invasiveness [2].
HCC impacts patients’ quality of life, and the most common symptoms are abdominal pain, weight loss, fever, and the worsening of hepatic synthetic function [4]. However, many patients are asymptomatic, and the diagnosis is made late when the disease is already advanced [5]. In addition, HCC, as with other cancers, is highly heterogeneous among patients due to genetic and epigenetic diversity and the tumor microenvironment [6], which makes treatment difficult. This heterogeneity is also reflected in the HCC immune response, which can be divided into some sub-classes, including “active immune”, “exhausted immune”, and “immune excluded” [7]. The “active immune” subtype associates with active T helper cells (CD4+) and CD8+ enrichment and responds well to treatment with immune checkpoint inhibitors (ICIs). The “exhausted immune” subtype is characterized by abundant TGFβ-secreting T lymphocytes, which are cells that show an exhausted status, and by the presence of immunosuppressive macrophages. In turn, the “immune excluded” subtype copes with increased an Treg cell number, has a worse prognosis, and does not respond to ICI therapies [1].
The main risk factors for HCC are infections by hepatitis B (HBV) and hepatitis C (HCV) viruses, alcohol abuse, obesity, and non-alcoholic steatohepatitis (NASH) [8]. NASH is part of a spectrum of liver disorders called non-alcoholic fatty liver diseases (NAFLD), which range from steatosis (NAFL), characterized by fat accumulation in liver tissue, to NASH, described as a pathological fat storage process associated with inflammation, liver injury, and fibrosis [9,10][9][10]. A “two-hit” model was proposed to explain NASH establishment [11]. The “first hit” is related to a dysregulated accumulation of lipids within liver tissue, generating hepatic steatosis, and the “second hit” triggers hepatocellular injury and inflammation due to liver oxidative stress and lipid peroxidation [11]. However, several studies have shown alternative mechanisms involving multiple pathways and metabolic hits that result in an unhealthy liver.
Within the NAFLD spectrum, only NASH, in association with fibrosis, can progress to cirrhosis and HCC [12,13][12][13]. Obesity is often present in patients with NASH, and this may increase the risk of developing HCC [14]. Additionally, studies have demonstrated that the gut microbiota plays crucial roles in nutrient harvest and fat storage, important processes for obesity progression [15], and deeply impacts NASH development, once patients show dysbiosis that favors alcohol-producing microbes and the occurrence of systemic inflammatory responses [16,17,18][16][17][18]. The connection between obesity-associated NASH pathways and HCC remains poorly understood. However, cumulative factors, such as inflammasome-mediated cytokines from obesity-associated NASH, are a possible link between these diseases with HCC development.
Obesity is characterized by a low-grade chronic inflammation [19] with dysregulation in the adipose tissues (ATs), among them white AT (WAT) and brown AT (BAT), endocrine organs that can modulate the inflammatory status through the secretion of several mediators, such as chemokines and cytokines [20]. The latter includes the pro-inflammatory molecules interleukin-1β (IL-1β) and IL-18, which are maturated by the action of assembled inflammasomes, multiprotein complexes that can also induce immunogenic pyroptotic cell death [21]. The inflammasomes are crucial for inflammatory homeostasis, directly influencing both obesity and NASH as well as liver cancer [22]. Although inflammasomes were first related to the organism’s defense against pathogens, studies have linked these molecules to many metabolic diseases, such as obesity [22], type 2 diabetes mellitus [16], NAFLD [16[16][23],23], and HCC [24].
2. Inflammasomes as a Mediator of Pro-Inflammatory Cytokines
Inflammation is a mechanism of the immune system to protect the host against pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Innate immunity recognizes PAMPs and DAMPs by germline-encoded pattern-recognition receptors (PRRs). Ligand recognition or cellular disorder is a trigger to activate downstream signaling pathways followed by the production of pro-inflammatory cytokines and chemokines [25]. There are some classes of PRRs, among them the Toll-like receptor (TLR), a transmembrane protein situated on the cell surface, absent in melanoma 2 (AIM2), a sensor for cytosolic DNA [26], and NOD-like receptor (NLR), which recognizes PAMPs in the cytoplasm [27,28][27][28]. In humans, there are 22 members in the NLR family, categorized into 4 subfamilies based on N-terminal domains: NLRA, with a domain formed by acidic transactivation; NLRB, accompanied by a baculoviral inhibitory repeat (BIR) domain; NLRC, containing a caspase-recruitment and activation domain (CARD); and NLRP, a pyrin-containing domain (PYD). In addition to that, NLRs have an intermediary NACHT, NOD, or NBS domain and a C-terminal leucine-rich repeat (LRR) domain [29,30][29][30]. Currently, the NLR family members, including NLRP1, NLRP3, and NLRC4, are best described as assembled inflammasome components, as well as AIM2 and pyrin [30]. In 2002, Martinon and colleagues showed that the inflammasomes are responsible for activating the protease caspase-1 [31]. Once activated, this protein cleaves zymogen forms of IL-1β and IL-18 into their active form [32]. In addition to activating these potent pro-inflammatory cytokines, caspase-1 is also capable of inducing pyroptotic cell death [33]. The connection between NLRs and caspase-1 occurs through the adaptor protein ASC (apoptosis-associated speck-like protein, containing a CARD), which contains an N-terminal PYD domain and a C-terminal CARD domain [34]. Studies have demonstrated that the absence of ASC in mice alters the maturation and release of IL-1β and IL-18 [35,36,37][35][36][37]. Therefore, ASC has an important role in the inflammasome function [37]. The canonical pathway of NLRP3 inflammasome activation and the consequent release of IL-1β and IL-18 is a process tightly controlled by two signals. The first involves TLR stimulation, which results in the transcriptional upregulation of genes encoding the inflammasome components, pro-IL-1β and pro-IL-18, via nuclear factor kappa B (NFκB) activity. The second signal, needed for activating caspase-1, is provided by microparticles, such as ATP, via the P2X7 receptor, reactive oxygen species, K+ efflux through ion channels, and cathepsin B activation [38,39][38][39]. In turn, the non-canonical pathway occurs through direct binding of LPS with caspase-11 (in mice) and caspase-4/5 (in humans), without TLR activation [40]. Then, active caspase-11 will cleave the IL-1β and IL-18 cytokines. Caspase-11 is also involved in the pyroptotic cell death [41]. The inflammasome-mediated cytokines can lead to an inflammatory microenvironment in obesity that contributes to NASH establishment and may support liver cancer development.3. IL-1β and IL-18 in Liver Cancer Progression
Inflammation has a dual effect in the cancer context, which can harm or benefit the tumor. Acute inflammation can trigger an anti-cancer immune response [42]. Chronic inflammation, on the other hand, can cope with augmented levels of cell growth and pro-angiogenic factors and with changes in the extracellular matrix that facilitate metastasis and DNA damage, thus contributing to an increase in cells with genetic alterations [43]. In the liver, IL-1β leads to, among other consequences, the release of IL-6 and TNF-α [44]. The increase in IL-6 levels is related to a worse prognosis in HCC patients [45]. In turn, IL-18 may influence the recruitment of T and NK cells [46]. There is a dysregulation in the NLRP3 inflammasome components in HCC depending on the stage of hepatocarcinogenesis [24]. Wei and others showed that during the development of HCC, the NLRP3 components are dysregulated depending on the disease stage, while in the inflammatory hepatic setting it copes with IL-1β and NLRP3 upregulation, and malignantly transformed liver cancer is downregulated [24]. IL-1β binds to interleukin-1 receptor type I (IL-1RI) and, after a series of cascades, activates NF-κB, which in turn is related to proliferation [47] and inflammation [48]. IL-1β can also promote the expression of the oncoprotein Gankyrin, which plays a critical role in HCC development and metastasis [49]. In 2019, Zong and colleagues demonstrated that M1 macrophages induced the expression of the programmed death ligand 1 (PD-L1) in HCC cells through IL-1β, supporting the pro-tumor role of M1 macrophages and IL-1β [50]. Corroborating with this study, another group recently showed that IL-1β induced PD-L1 expression in HCC, a phenomenon that contributes to tumor immune resistance in HCC [51]. Polymorphisms in the IL-1 family genes were described in HCC patients, which suggests that IL-1β contributes to HCC susceptibility and plays an important role in the progression of this neoplasm [52]. Another inflammasome-mediated cytokine, IL-18, is also involved in liver cancer occurrence. This cytokine is upregulated in HCV patients, an important risk factor for HCC. The studies showed that the IL-18 receptor (rhIL-18) is expressed in both HCC patients and cell lines [53]. The activation of this receptor was capable of increasing NF-κB activation and anti-apoptotic molecule expression, including Bcl-xL and xIAP [53]. Therefore, the expression of rhIL-18 and an antiapoptotic mechanism involving NF-κB activation in HCC cells may be related to poor prognosis in HCC patients [53]. Corroborating with these findings, a 2020 study indicated that IL-18 single nucleotide gene polymorphism could be a marker for HCC in patients with HCV-related cirrhosis [54], and IL-18 gene polymorphisms could be used as a potential non-invasive diagnostic tool for HCC patients at early stages [55], which is supported by recent findings that IL-18 high levels are found in HCC patients with poor prognosis [56]. In contrast, studies have also demonstrated a dual function of IL-18 in tumor progression. IL-18 can regulate Th17 cells in vitro and in vivo in the HCC model [57]. Th17 cells can both worsen prognosis [58,59][58][59] and affect antitumor cytotoxicity by CD8+ T-cells [59,60][59][60]. In human hepatocytes, it was demonstrated in vitro that IL-18 inhibited HBV replication but also promoted HepG2 cell metastasis and migration [59]. Taking these results, the IL-1β and IL-18 effects on HCC are demonstrated in Figure 1.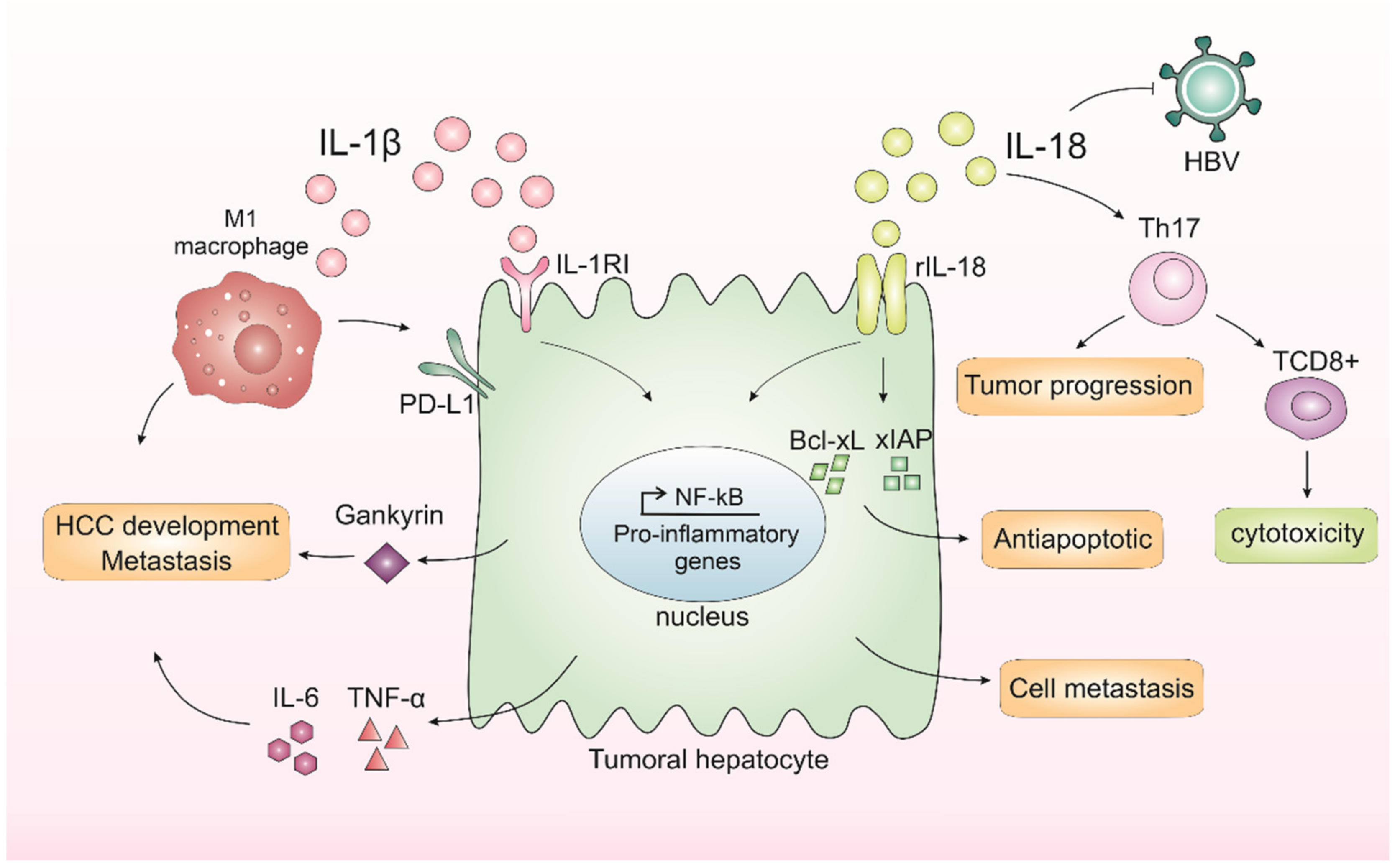
Figure 1. Inflammasomes-mediated cytokines IL-1β and IL-18 in liver cancer progression. In the liver, IL-1β leads to the release of IL-6 and TNF-α, cytokines related to a worse prognosis in hepatocellular carcinoma (HCC). IL-1β binds to interleukin-1 receptor type I (IL-1RI) and, after a series of cascades, activates nuclear factor kappa B (NF-κB), which in turn is related to proliferation and inflammation. IL-1β is also capable of promoting the expression of an oncoprotein, Gankyrin, which plays a critical role in HCC development and metastasis. M1 macrophages induce the expression of programmed death ligand 1 (PD-L1) in HCC cells through IL-1β, supporting the pro-tumor role of M1 macrophages and IL-1β. IL-18 receptor (rIL-18) is capable of increasing NF-κB activation and anti-apoptotic molecule expression, including Bcl-xL and xIAP. IL-18 can also regulate Th17 cells, which can both worsen prognosis or affect antitumor cytotoxicity by CD8+ T-cells. Finally, IL-18 could inhibit HBV replication but could also promote hepatocyte cell metastasis and migration.
References
- Llovet, J.M.; López, S.A.; Fajes, J.L.H.; Martín, L.C. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236.
- Shen, S.; Lin, Y.; Yuan, X.; Shen, L.; Chen, J.; Chen, L.; Qin, L.; Shen, B. Biomarker MicroRNAs for Diagnosis, Prognosis and Treatment of Hepatocellular Carcinoma: A Functional Survey and Comparison. Sci. Rep. 2016, 6, 38311.
- Di Bisceglie, A.M. Epidemiology and Clinical Presentation of Hepatocellular Carcinoma. J. Vasc. Interv. Radiol. 2002, 13, 169–171.
- Balogh, J.; Balogh, J.; Victor, D., III; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53.
- Cajal, S.R.Y.; Capdevila, C.; Hernandez-Losa, J.; De Mattos-Arruda, L.; Ghosh, A.; Lorent, J.; Larsson, O.; Aasen, T.; Postovit, L.-M.; Topisirovic, I. Cancer as an ecomolecular disease and a neoplastic consortium. Biochim. Biophys. Acta-Rev. Cancer 2017, 1868, 484–499.
- Giraud, J.; Chalopin, D.; Blanc, J.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697.
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750.
- Brunt, E.M. Non Alcoholic Steathohepatitis (NASH). Semin. Liver Dis. 2004, 24, 3–20.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two ‘Hits’? Gastroenterology 1998, 114, 842–845.
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28, 64–70.
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285.
- Saitta, C.; Pollicino, T.; Raimondo, G. Obesity and liver cancer. Ann. Hepatol. 2019, 18, 810–815.
- Rosenbaum, M.; Knight, R.; Leibel, R.L. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol. Metab. 2015, 26, 493–501.
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185.
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397.
- Brestoff, J.R.; Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 2015, 161, 146–160.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188.
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 27, 1045–1059.
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62.
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837.
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR Gene Family: A Standard Nomenclature. Immunity 2008, 28, 285–287.
- Dagenais, M.; Skeldon, A.; Saleh, M. The inflammasome: In memory of Dr. Jurg Tschopp. Cell Death Differ. 2012, 19, 5–12.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426.
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice Deficient in IL-l beta-Converting Enzyme Are Defective in Production of Mature IL-lp and Resistant to Endotoxic Shock. Cell 1995, 80, 401–411.
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298.
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574.
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218.
- Andersen, K.; Eltrich, N.; Lichtnekert, J.; Anders, H.J.; Vielhauer, V. The NLRP3/ASC inflammasome promotes T-cell-dependent immune complex glomerulonephritis by canonical and noncanonical mechanisms. Kidney Int. 2014, 86, 965–978.
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40.
- Guo, H.; Callaway, J.; Ting, J.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677.
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121.
- Zhang, Z.; Shao, X.; Na Jiang, N.; Mou, S.; Gu, L.; Li, S.; Lin, Q.; He, Y.; Zhang, M.; Zhou, W.; et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 2018, 9, 983.
- Liu, X.; Yin, L.; Shen, S.; Hou, Y. Inflammation and cancer: Paradoxical roles in tumorigenesis and implications in immunotherapies. Genes Dis. 2021, 1–14.
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41.
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102.
- Porta, C.; DE Amici, M.; Quaglini, S.; Paglino, C.; Tagliani, F.; Boncimino, A.; Moratti, R.; Corazza, G.R. Circulating interleukin-6 as a tumor marker for hepatocellular carcinoma. Ann. Oncol. 2008, 19, 353–358.
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72.
- Lin, A.; Karin, M. NF-kappaB in cancer: A marked target. Semin. Cancer Biol. 2003, 13, 107–114.
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734.
- Su, B.; Luo, T.; Zhu, J.; Fu, J.; Zhao, X.; Chen, L.; Zhang, H.; Ren, Y.; Yu, L.; Yang, X.; et al. Interleukin-1β/Iinterleukin-1 receptor-associated kinase 1 inflammatory signaling contributes to persistent Gankyrin activation during hepatocarcinogenesis. Hepatology 2015, 61, 585–597.
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 macrophages induce PD-L1 expression in hepatocellular carcinoma cells through IL-1β signaling. Front. Immunol. 2019, 10, 1643.
- Numata, Y.; Akutsu, N.; Ishigami, K.; Koide, H.; Wagatsuma, K.; Motoya, M.; Sasaki, S.; Nakase, H. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem. Biophys. Rep. 2022, 30, 2405–5808.
- Tak, K.H.; Yu, G.I.; Lee, M.Y.; Shin, D.H. Association between polymorphisms of interleukin 1 family genes and hepatocellular carcinoma. Med. Sci. Monit. 2018, 24, 3488–3495.
- Asakawa, M.; Kono, H.; Amemiya, H.; Matsuda, M.; Suzuki, T.; Maki, A.; Fujii, H. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int. J. Cancer 2006, 118, 564–570.
- Eldesoky, A.A.; Ahmed, N.A.F.; Zaghloul, H.E.; Aziz, A.A.A. Interleukin-18 polymorphism as a diagnostic tumor marker for hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Egypt Liver J. 2020, 10, 51.
- Sharafelldin, H.; Mors, A.; Elghobary, H.; Osman, E.; Rady, N. Association between TNF-α, Interleukin-18 Polymorphisms and Risk of Hepatocellular Carcinoma in Egyptian patients. Asian Pac. J. Cancer Prev. 2021, 22, 887–891.
- Yao, Z.; Zhao, M.; Gao, G.; Yang, J.; Wang, Z.; Liu, Y. Prognostic Role of IL-18 in Various Human Cancers and Radiation Injuries: A Meta-Analysis. Dose-Response 2020, 18, 1–7.
- Markowitz, G.J.; Yang, P.; Fu, J.; Michelotti, G.A.; Chen, R.; Sui, J.; Yang, B.; Qin, W.-H.; Zhang, Z.; Wang, F.-S.; et al. Inflammation-dependent IL18 signaling restricts hepatocellular carcinoma growth by enhancing the accumulation and activity of tumor- infiltrating lymphocytes. Cancer Res. 2016, 76, 2394–2405.
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.-X.; He, H.-W.; Cai, X.-Y.; Zhou, J.; Cheng, Y.-F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3.
- Zhang, Y.; Li, Y.; Ma, Y.; Liu, S.; She, Y.; Zhao, P.; Jing, M.; Han, T.; Yan, C.; Wu, Z.; et al. Dual effects of interleukin-18: Inhibiting hepatitis B virus replication in Hepg2.2.15 cells and promoting hepatoma cells metastasis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, 565–573.
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol. Cancer 2011, 10, 150.
More