Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Marco Kruppa and Version 3 by Lindsay Dong.
Marine natural products are a source of essential significance due to a plethora of highly diverse biological properties. The naturally occurring (aza)indole alkaloids variolin B (1), meridianins (2), and their synthetic hybrids meriolins (3) exhibit potent kinase inhibitory activities.
- indole alkaloids
- marine natural products
- natural product syntheses
- variolins
- meridianins
- meriolins
1. Syntheses of Variolins
1.1. First Total Synthesis by Morris and Anderson
The first total synthesis of variolin B (1) was achieved by Morris and Anderson in 2001 [1]. Later in 2005, they published the full details of their synthetic strategy together with the synthesis of the synthetic analog desoxyvariolin B [2]. They recognized the C2-symmetry of intermediate 8, which is cyclized to the pyridopyrrolopyrimidine in the following key step. After halogen lithium exchange in the methylthiopyrimidine 4, the reaction with diethyl carbonate (5) gave the symmetric ketone 6. The reaction with the lithiated pyridine 7, followed by the key step tandem deoxygenation and cyclization in the presence of triethylsilane and TFA led to the variolin core structure 9. The introduction of the amino groups was achieved by oxidizing the dimethylthiol 9 with m-chloroperbenzoic acid (mCPBA) to the corresponding disulfoxide, which was reacted with p-methoybenzylamine (PMB amine) (10) to give the bisprotected amine 11. Demethylation of 11 and removal of the PMB protecting groups gave the trifluoroacetate salt of the title compound, which was neutralized with concentrated ammonia to give variolin B (1) in an eight-step synthesis and an overall yield of 11% (Figure 14) [1].
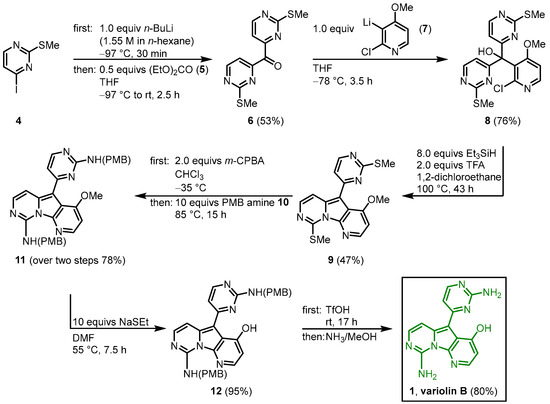
Figure 14.
First total synthesis of variolin B (
1
) by Morris and Anderson
[1]
.
1.2. Synthesis by Molina and Fresneda
The next synthetic approach was conducted by Molina and Fresneda, who published their syntheses of 1 in 2002 [3] and a modified synthetic route together with the synthesis of an analog in 2003 [4]. This approach starts with the synthesis of the 7-azaindole 16. Aldehyde 13 was condensed with azidoacetate 14 and the resulting vinyl azide 15 cyclized to azaindole 16 via a nitrene insertion. After N-protection with 2-(trimethylsilyl)ethoxymethyl (SEM), the chloride key intermediate 19 was synthesized in a two-step procedure (Figure 25).
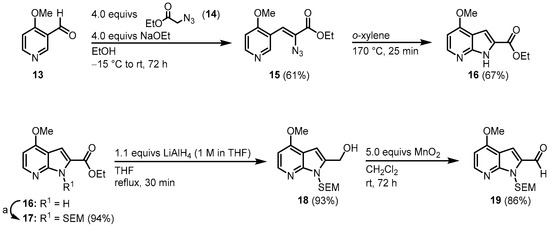
Figure 25. Synthesis of key intermediate azaindole 19. Reaction conditions for a: first: 1.4 equivs NaH, DMF, rt, 45 min. Then: 1.4 equivs SEM-Cl, rt, 12 h [3].
Next, two different approaches are reported (Figure 36). Aldehyde 19 was similarly condensed as aldehyde 13 to give vinyl azide 20. After N-SEM-deprotection, a Staudinger reaction with triphenylphosphane led to iminophosphorane 21 in a one-pot reaction. Reaction with benzyl isocyanate (22) in the key aza-Wittig reaction gave a non-isolable carbodiimide that subsequently cyclized to the desired pyridopyrrolopyrimidine moiety 23.
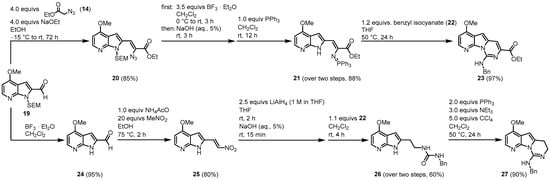
Figure 36.
Two approaches to the synthesis of the tricyclic pyridopyrrolopyrimidine structures,
23
and
27
[3]
.
Molina and Fresneda developed a second approach to obtain the tricyclic variolin core without the ester group at C-7. After the N-SEM-deprotection of 19, a nitroaldol condensation with nitromethane led to the formation of 25. Treatment with lithium aluminum hydride gave the corresponding 2-(2-aminoethyl)-7-azaindole, which was sequentially converted to the urea derivative 26 with benzyl isocyanide (22) without isolation. The 26 was dehydrated to the carbodiimide, which subsequently cyclized to the dihydropyrimidine 27 using the Appel reagent (CCl4/PPh3/NEt3). Applying both synthetic approaches, an oxygen substituent is placed at C-4 and a nitrogen substituent at C-9. The next step was to introduce the 2-aminopyrimidine ring at C-5, consequently leading both approaches to the acylated intermediate 31. The reaction of 23 with phosphorus oxychloride and N,N-dimethylacetamide (DMA) (28) allowed the direct introduction of an acetyl group at C-5. Ester hydrolysis led to the carbonic acid 30, and the thermal treatment forced the formation of intermediate 31 by decarboxylation. The route starting from 27 began with the introduction of a bromine substituent at C-5 and the reaction of bromine 32 with n-tributyltin(1-ethoxyvinyl)stannane (33) in the presence of dichlorobis (triphenylphosphine)-palladium(II) introduced to the acetyl group at C-5. Oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave the intermediate 31 (Figure 47).
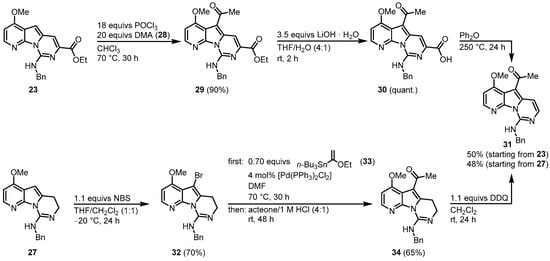
Figure 47.
Introduction of an acetyl group at C-5
[3]
.
The 2-aminopyrimidine substituent was synthesized using a protocol developed by Bredereck (Figure 58) [5]. Enaminone 36 was synthesized from 31 with N,N’-dimethylformamide di-tert-butylacetal (35) in DMF. Condensation with guanidine hydrochloride (37) led to ring closure and formed the desired 2-aminopyrimidine 38.
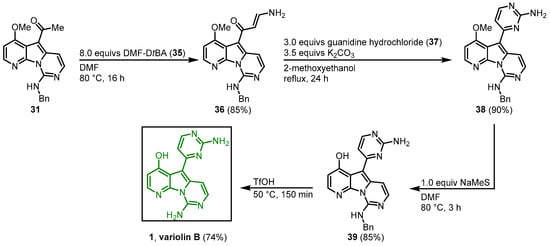
Figure 58.
Synthesis of the 2-aminopyrimidine ring to give access to variolin B (
1
)
[3]
.
1.3. Variolin B Approach by Alvarez
In 2003, Alvarez published the synthesis of variolin B (1) and the synthetic analog desoxyvariolin B [6][7][8]. Starting from 4-methoxy-7-azaindole (40), a lithium carboxylate was used as an N-protecting group as well as an ortho-directing substituent to form a 2-lithio-7-azaindole with a protocol by Katritzky [9]. Reaction with 2-(1,3-dioxoisindolin-2-yl)acetaldehyde (41) gave the alcohol 42 that was protected with dihydropyran. N-deprotection of 43 by hydrazinolysis gave the aminoacetal 44. Ring closure was achieved by the reaction with N-tosylcarbonimidic dichloride (45) and diisopropylethylamine (DIPEA) giving 46 in a diasteriometric mixture in a ratio of 1:1. Removal of the O-tetrahydropyran (THP) protecting group and elimination of the resulting hydroxy group by the formation of its mesylate and treatment with triethylamine afforded the pyridopyrrolopyrimidine scaffold (48). Regioselective iodination with N-iodosuccinimide (NIS) gave the key intermediate 49 (Figure 69).
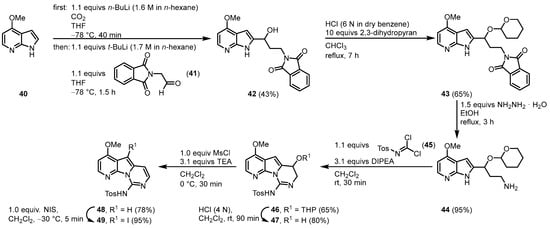
Figure 69.
Synthesis of the key intermediate iodide
49
[6]
.
A Stille reaction of 49 and 2-acetylamino-4-trimethylstannylpyrimidine (50) in the presence of tris(dibenzylideneacetone)dipalladium(0) afforded 51. The O-demethylation and N-acetyl-deprotection were achieved by the treatment of 51 with hydrobromic acid, and after reductive photolysis with hydrazine as a reducing agent and 1,4-dimethoxybenzene as an electron source, the tosyl group was cleaved to give variolin B (1) in a 10-step synthesis with an overall yield of 1% (Figure 710).
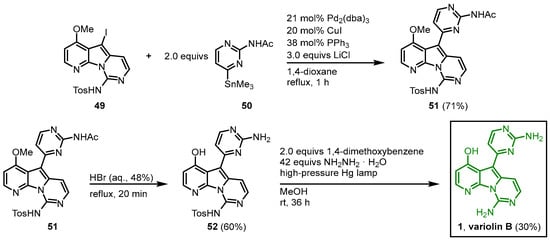
Figure 710.
Synthesis of variolin B (
1
) via Stille coupling as a key reaction step
[6]
.
1.4. Synthesis of Variolin B by Burgos and Vaquero
The 2008 approach by Burgos and Vaquero to synthesize variolin B (1) followed the strategy to design the highly functionalized trihalo-substituted pyridopyrrolopyrimidine core 55 and introduce the substituents via palladium-mediated cross-coupling reactions [10][11]. The functionalized 7-azaindole 53 was synthesized from 7-azaindole in six single steps [12]. The 53 was reacted with N-tosylmethyl dichloroformimide (54) under phase-transfer conditions in the two-phase system LiOH (aq., 30%)/CH2Cl2 (1:1) with tetrabutylammonium chloride to give the trihalo-substituted compound 55. The C-9 amino substituent was introduced by a palladium-mediated C-N bond formation, using lithium bis(trimethylsilyl)amide (LiHMDS) and triphenylsilylamine as an ammonia source. The reaction required the use of the ligand [1,1′-biphenyl]-2-yldi-tert-butylphosphane (JohnPhos). After N-acetyl-protection, 56 was obtained (Figure 811).
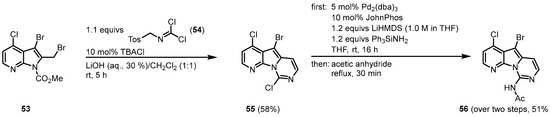
Figure 811.
Synthesis of the trihalo core and introduction of the C-9 amino substituent
[10]
.
Next, in a debromination-iodination process, tris(trimethylsilyl)silane (TTMSS) and azobisisobutyronitrile (AIBN) and subsequently NIS were used to exchange the bromo compound 56 to the more reactive iodo derivative 57. In a palladium-catalyzed cross-coupling reaction with the pyrimidyl stannyl reagent 58, the C-C bond at C-5 was formed and the deprotection of both amino groups led to 59. Then, in a palladium-promoted C-O coupling microwave (MW) reaction with sodium tert-butoxide, the tert-butyl group was introduced at C-4 to give the tert-butyl ether 60, and in a final step, the tert-butyl moiety was cleaved to give variolin B (1) (Figure