Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ricardo Lagoa and Version 3 by Conner Chen.
Staphylococcus aureus is a versatile pathogen known to cause hospital- and community-acquired, foodborne, and zoonotic infections. As a multi-faceted pathogen, it is essential to consolidate the knowledge on its pathogenesis, including the mechanisms of virulence regulation, development of antimicrobial resistance and biofilm formation, to make it more amenable to classical and novel therapeutic strategies such as those based on nanomaterials. The present overview focuses on the impact of S. aureus on human health and the role of virulence factors and biofilms during pathogenesis.
- Staphylococcus aureus
- virulence
- immune response
- biofilm
- two-component systems
- nanotherapeutics
1. Staphylococcus aureus and Its Impact on Human Health
S. aureus, a normal flora of skin and mucosa, seizes the opportunity to cause opportunistic infections, most commonly from superficial to invasive infections like skin infections, bacteremia or pneumonia, etc., either as nosocomial or community infections [1][3]. The evolution of drug-resistant S. aureus, especially methicillin-resistant S. aureus (MRSA) report in the late 1990s [2][17] and vancomycin-resistant S. aureus (VRSA) in 2002 [3][18], demanded careful attention as it complicates the treatment. The WHO revealed in 2014 that 86% of clinical S. aureus strains were resistant to methicillin [4][19]. Frequent use of certain antibiotics in the hospital environment has promoted the emergence of multidrug-resistant (MDR) strains. Regulation of significant virulence factors and formation of tolerant or persistent subpopulations (persister cells) also adds to the risk of invasive and recurrent infections by S. aureus [5][20]. The cellular expression of surface proteins facilitates bacterial adhesion to the host cell or tissue, thereby causing adverse pathogenesis in humans [6][7][21,22]. These proteins are particularly specified to promote bacterial adhesion to the host tissues. Several structural and secreted virulence factors in S. aureus play a crucial role during pathogenesis (Table 1).
Table 1.
Critical virulence processes and associated genes in
| Virulence Processes | Virulence Factors Involved | Responsible Genes |
|---|---|---|
| Attachment | Microbial surface components recognizing adhesive matrix molecules involving surface proteins such as bone sialoprotein-binding protein, fibronectin-binding proteins, clumping factors, and fibrinogen-binding protein | bbp, fnbA, fnbB, clfA, clfB, sdrD, sasG, fib, and cna |
| Invasion or tissue penetration | Enzymes able to break down lipids, phospholipids, proteins (elastin), DNA, and hyaluronic acid | hysA, nuc, gehB, plc, sepA, sspA, and V8 |
| Destroying or evading host immune system | Pore-forming toxins like leukocidins, phenol-soluble modulins, protein A, CHIPS, Eap, staphyloxanthin, staphylococcal complement inhibitor, and capsular polysaccharides | lukS-PV, hlg, lukF-PV, crtN, spa, psm-a gene cluster, chp, scn, eap, cap5, and 8 gene clusters |
| Persistence and tolerance | Factors involved in intracellular persistence and biofilm formation such as polysaccharide intracellular adhesions, and formation of small colony variants | ica operon, dnaK, and hemB mutation |
| Toxins mediated infections and sepsis | Toxic shock syndrome toxin-1, α-toxin, enterotoxins, exfoliative toxins A and B, and lipoteichoic acid | sea, hla, tstH, eta, and etb |
2. The Host Immune Response to Staphylococcus aureus
The host immune response elicits the defense machinery against bacterial invasion. Upon bacterial cell invasion, the host immune system instantly brings forth the trapping mechanism by recognizing the antigenic determinant on the pathogen. In addition, the early response signals to other immune cells to localize to the site of invasion or infection. Following pathogen- and host-derived chemotactic factors, host immune cells like neutrophils from the bone marrow or peripheral blood migrate to the site of infection. The host system triggers polymorphonuclear leukocytes (PMNs), which probably stand as the first line of defense. However, there may be slight alterations in PMN response against biofilm-forming S. aureus [9][10][24,25].
Nevertheless, the host pro-inflammatory response at the early stage, which is not considered very effective, leads to the activation of PMNs (i.e., neutrophils/granulocytes) and thereby the release of lactoferrin, elastase, and other relevant components with antimicrobial potential [10][25]. While the activation of PMNs triggers the localized influx of immune system players at the infection site, it also promotes opsonization with the aid of IgG to eliminate S. aureus [9][24]. In parallel, PMNs also activate phagocytosis to eradicate the free-living planktonic cells by reactive oxygen species (ROS) generation [11][26], and has a significant role in reducing the biofilms of S. aureus. In addition to ROS, the degranulation process (fusion of azurophilic and specific granules with phagosomes) enriches the vacuole lumen of phagosomes containing S. aureus with antimicrobial peptides and proteins to ensure bacterial clearance [12][27]. The azurophilic and other granules comprise enzymes, cytokines, antimicrobial peptides, and other components such as α-defensins, proteinase-3, elastase, cathepsins, azurocidin, lysozyme, myeloperoxidase, etc. [13][28].
The study by Alegre and co-workers documented the immune response in skin and soft tissue infections and hypothesized that their occurrence in healthy persons may be due to a suppressed immune system compared to uninfected persons [14][29]. On the contrary, the experimental analysis indicated that developing systemic immune responses leads to an elevated level of innate cytokines in infected persons. The same work highlighted the production of interleukins IL-17, IL-22, and IL-10 by macrophages. Further, IL-6 can indicate T lymphocyte differentiation, which promotes Th17 effector or memory cells. Another unique mechanism from neutrophils has been suggested by Brinkmann et al. [15][30], in which they secrete structures constituting chromatin in a de-condensed form along with histones and cytosolic and azurophilic granule proteins, that effectively act against S. aureus. Moreover, staphylococcal toxins stimulate Th1/Th17 responses leading to asthma and allergic rhinitis [16][17][31,32].
3. Immune Evasion by Staphylococcus aureus
S. aureus elicits a series of proinflammatory responses to avoid the actions of the host immune system. For instance, protein A and immunoglobulin-binding protein A (Sbi) of S. aureus directly bind the Fc portion of IgG to avoid opsonization (antibody-mediated killing) as well as phagocytic activities [18][33]. Further, S. aureus Coa (coagulase) induces the conversion of fibrinogen into fibrin, which assists in protecting the bacterial cell from the host. Peripheral monocytes in the blood promote the release of IL-8, which trigger neutrophil activation. S. aureus, however, can induce the synthesis of ClfA (clumping factor A) and SdrE (serine aspartate repeat protein). These proteins prevent the regulatory proteins Factor I and Factor H from activating complement pathways, which cleave the Cd3 domain and block complement pathway-mediated phagocytosis [19][20][34,35]. While analyzing the role of complement in S. aureus bacteremia, several patients were found with low levels of C3 and C4, pointing to the activation of the classical pathway. However, there were indications of activating the alternative pathway to eliminate the pathogen [21][36]. In some instances, protein A from S. aureus binds to IgA and IgM, preventing complement binding and obviating the classical pathway of complement defense. Hypervirulent S. aureus mainly disrupts the overall host defense by influencing antimicrobial peptides, phagocytic elimination, and infection resolution [22][37]. Likewise, S. aureus produces several proteins like the staphylococcal complement inhibitor, chemotaxis inhibitory protein of Staphylococcus, extracellular adherence protein, hemolysins, Staphylococcal binding of IgG, aerolysin, phenol-soluble modulins, nuclease, leukocidins, FPR2 inhibitory proteins, and staphylococcal superantigens among others, to evade host immune mechanisms like neutrophil extravasation, priming, chemotaxis, activation of neutrophils, opsonization and phagocytosis, NET formation, and bacterial killing by neutrophils [23][38].
4. Challenging Issues of Staphylococcus aureus in Healthcare
The more problematic issues related to S. aureus infections are detailed below. The outbreak of MRSA has had a vast impact over the years, in a particular assortment from hospital- to later community-based infections, in persons lacking medical attention [5][24][20,39]. Nevertheless, in recent decades, MRSA has shown resistance to the extended spectrum of beta-lactam (ESBL) antibiotics, including penicillins, cephalosporins, and carbapenems [25][40]. However, 80% of the mortality rate in hospitalized infections is due to biofilm formers [1][3]. The recurrent infection range of MRSA comprises skin and soft tissue infections, bacteremia, endocarditis, urinary tract infection, osteomyelitis, nasal colonization, and cystic fibrosis complications [5][20]. In addition, MRSA also dominates the list of implant-associated infections [26][41].
The more typical S. aureus illness is skin and soft tissue infection, which may include impetigo and purulent cellulitis [6][21]. Impetigo is common in staphylococcal infections, particularly in the crusty lesion of extremities [14][29]. Toxic shock syndrome occurs due to toxic shock syndrome toxin [27][42], primarily caused by the use of absorbable tampons, and it includes serious clinical conditions, such as multi-organ coupled septic shock. Considering the crucial role of S. aureus as an inhabitant of nares, Nurjadi et al. [28][43] underlined the connection between pvl, found among intercontinental travelers, and the lukL/lukS genes associated with the severity of skin and soft tissue infections. MRSA has been reported as a major cause of healthcare-associated pneumonia in the statistical analysis performed by Walter et al. [29][44] in European countries. Next to Pseudomonas aeruginosa, S. aureus is a major colonizer of the lungs in patients with cystic fibrosis. S. aureus influences cystic fibrosis transmembrane conductance regulator protein present in the epithelia. It leads to mucus accumulation in the respiratory tract, resulting in dyspnea and ultimately to the morbidity and mortality of cystic fibrosis patients [30][45]. Endotracheal tubes are the primary source of acquiring pneumonia by exhaustive use of ventilators in intensive care unit patients through the development of biofilms [31][46]. Pacemakers, defibrillators, and heart valve implants are other targets for S. aureus cardiovascular infections, which are the more common causes of early-onset prosthetic valve endocarditis [32][47].
Notably, sepsis and endocarditis producing an elevated mortality rate was observed in various vascular catheters [1][3]. Fibrinogen-binding proteins, such as clumping factors (Clfs) ClfA and ClfB, in addition to SdrE, result in platelet aggregation, which paves the way for endocarditis [33][48]. Previous research identified S. aureus as the second most common species in ventricular shunt infection [34][35][49,50]. Intracranial pressure and meningeal irritation were major symptoms described in cerebrospinal fluid shunt infections [36][51]. An increased risk of shunt infection was observed after the reimplantation of shunts and cerebral spinal fluid leakage in a post-operative crisis. Furthermore, S. aureus attempts to cause a viscous infection called osteomyelitis in the case of bones and joints [37][52]. Another current concern with S. aureus is infections during orthopedic procedures, especially knee or hip arthroplasty [38][53].
Urinary tract infections by S. aureus are rare. Yet, conditions like older age, hospital exposure, urologic surgical procedures, long-term urinary tract catheterization, urinary tract obstruction, and malignancy favor S. aureus-associated hematuria, dysuria, bacteriuria, or bacteremia [39][54]. The study by Gjodsbol et al. [40][55] indicated that S. aureus is present in more than 80% of chronic wound infections typified by diabetic foot ulcers, venous ulcers, and pressure sores.
5. Biofilm Formation in Staphylococcus aureus
A crucial biological tactic that enables bacteria to resist attacks from the host and other threats in a hostile environment is the production of three-dimensional structures termed ‘biofilms’ [41][42][56,57]. S. aureus engages itself in the formation of biofilms on distinct surfaces and play significant roles in chronic infections in humans [43][58]. S. aureus prevails in biofilm formation on biotic (e.g., host tissue) or abiotic substrata, especially indwelling biomedical devices. To date, S. aureus has been reported on various biomedical materials, such as endotracheal tubes, dental prostheses or implants, intravenous catheters, vascular prostheses, intrauterine devices or urinary catheters, cardiac pacemakers, contact lenses, prosthetic joints, and prosthetic heart valves [44][8].
5.1. Overview of Biofilm Formation
To produce biofilms in a biological niche, the population of S. aureus follows a sequence of steps: (i) adhesion in a reversible state; (ii) irreversible microcolonization; (iii) propagation of 3D biofilm; (iv) maturation; and (v) dispersion [45][46][59,60] (Figure 1). Initial adhesion on the substrate is established by a group of free planktonic bacterial cells. The hydrophobic and van der Waal’s forces facilitate the initial adhesion to the substratum. This non-covalent interaction between the bacterial cells and the substrate is initially reversible. Therefore, the film formed by the early attachment always appears to be fragile. Subsequently, the adhesion becomes irreversible with stronger binding of the bacteria to the substrate. This is made possible as the bacterial population imparts charges on the surface to which it is attached. The conditioning of the film enables the consequent synthesis of specific adhesive proteins and lipopolysaccharides from the S. aureus inhabitants. In addition, flagella and pili also play an indispensable role. Quorum sensing is the pioneering machinery followed by the bacterial cells, allowing them to form biofilms via distinct gene expression, and communication among the individual cells of a population (for instance, bacterial cells in biofilm) that relies on signaling molecules described as autoinducers (AI) [45][59].
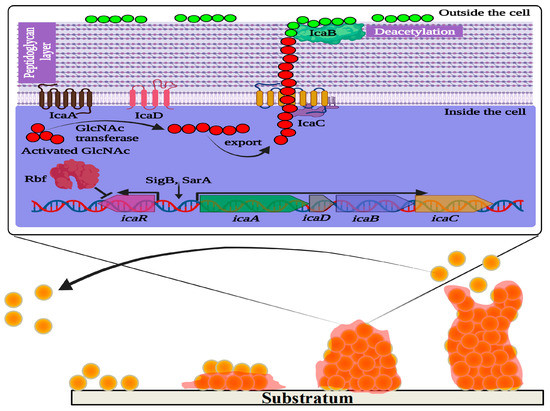
Figure 1. Schematic representation of ica operon regulating polysaccharide intercellular adhesin (PIA)-dependent biofilm formation in Staphylococcus aureus. IcaA and IcaD are two transmembrane proteins involved in the synthesis of N-acetylglucosamine (GlcNAc) oligomers that are 15–20 residues in length from activated precursor UDP-GlcNAc units. The growing GlcNAc chain is likely to be exported by another membrane protein named IcaC, which is also suspected to modify PIA molecules. IcaB is an outer membrane protein that generates positive charges in the GlcNAc polymer molecules by deacetylation. The cationic character of PIA is critical for its role in attachment to the anionic bacterial cell surface. IcaR is a repressor protein that controls the expression of icaADBC. Rbf promotes biofilm formation by regulating the expression of icaR. In the lower panel of the figure, the four main stages of biofilm formation are depicted: adhesion, colonization, maturation, and dispersion.
5.2. Biofilm Matrixome and Dynamics
The stacking of bacterial cells is accomplished by the expression of numerous virulent genes that produce essential components of the biofilm, including nucleic acid (eDNA), proteins, lipids, and polysaccharides that constitute the extracellular polymeric substances (EPS) matrix [46][60]. As the biofilm matures, the accumulation of components in the EPS matrix consolidates the three-dimensional structure, including its thickness and strength. The multilayered mature biofilm architecture maintains the hierarchy in which the deeper zone encompasses closely packed bacterial cells and persister cells, while the upper zone is more sparsely populated. The persister cells (~1% of the biofilm population) residing in the biofilm tolerate the antibiotics in a dormant or quiescent state, and such action diverges from actual drug resistance [47][48][61,62]. The eDNA in biofilm has a vital role in establishing viscoelasticity control, aiding the defensive role of biofilm.
There may be specific channels for the transport of water and nutrients to carry out a low rate of metabolism. Nonetheless, the cells in the deeper region typically remain dormant, possibly because of insufficient nutrients and oxygen that are depleted at the upper region of the biofilm [49][63]. Four distinctive states of bacterial cells have been identified in biofilms: aerobic cells, fermentative cells, slow growers/dormant cells, and dead cells [49][63]. Dead cells are also considered the most crucial source of eDNA that facilitates the live bacterial cells to adhere to the surface [50][64]. The physiological changes in the cells residing in biofilms (e.g., less motility and hydrophobicity) resist the action of antimicrobials and other stress conditions, like pH, osmotic pressure, chemicals, desiccation, nutrition, temperature, radiation, etc. [46][60]. Further, the EPS matrix hinders the diffusion/penetration of antibiotics inside the biofilm. Apart from these, active efflux pumps and the divergent expression of certain genes configuring membrane transport have been described as contributing to resistance to various antibiotics [51][65]. Finally, in the biofilm dispersion stage, cells residing in the biofilm produce proteases and nucleases to degrade the biofilm matrix. Once the biofilm matrix degrades, the cells leave the biofilm and enter the planktonic stage again to initiate the adhesion on another surface or continue their life cycle as free planktonic cells to infect other body parts [43][58]. The biofilm dispersion may be mechanical, through a fluid run of the host, or genetically programmed [52][66]. The following section discusses distinct genes involved in biofilm formation and their contribution in S. aureus virulence.
Bacterial cells undergo unique gene transfer mechanisms within the biofilm through conjugation, transformation, and transduction [53][67]. With horizontal gene transfer mechanisms, the resistance gene(s) prevailing in a single bacterium can be transferred to other cells. Thus, biofilm formation enables the development of antibiotic-resistant strains through the regulated production of the EPS matrix and the horizontal transfer of antibiotic-resistance genes.
6. Virulence Regulation in Staphylococcus aureus
6.1. Regulation of the Two-Component System AgrAC in
Staphylococcus aureus
Heterogeneous gene expression has been found in S. aureus to regulate its virulence in most conditions. Previous studies assessed gene regulation contributing to the virulence pattern in S. aureus, for example, those that are induced by low levels of antibiotics [54][68]. Production of virulence factors and biofilm formation in S. aureus is responsible for causing a wide range of infections. Many of them are controlled by the quorum sensing (QS) system in S. aureus, named the accessory gene regulator (agr) system. QS allows gene expression that regulates the physiological activity of an individual/population, promoting pathogenesis and antibiotic resistance.
Bronesky et al. (2016) have described that the induction of virulence factors in S. aureus is regulated by the complex of genes in an agrACDB cassette possessing two signaling transducing modules, a receptor histidine kinase (HK) agrC, and a cytoplasmic response regulator (RR) agrA, as represented in Figure 2 [55][69]. The agrC-agrA modules form a classical two-component system (TCS) composed of an HK sensor and a RR protein, important in bacterial responses to environmental factors, such as antibiotics and QS signals [56][70]. The locus agrD encodes the precursor of autoinducing peptide (AIP), and AgrB functions as an endopeptidase and chaperone protein involved in the maturation and release of active AIP. The agr system gets activated upon reaching the threshold level of AIPs in the external environment. Upon binding with AIPs, AgrC phosphorylates AgrA, which binds to the region between the agr promoters, activating both P2 and P3 for the synthesis of RNAII (agrA-D) and RNAIII transcripts, respectively (Figure 2). The transcription factor agrC thus completes a strong positive feedback loop and upregulates the expression of α-phenol-soluble modulin (PSM) and β-PSM proteins. The hairloops of RNAIII upregulate or suppress target genes by offering transcriptional/translation control. For instance, RNAIII binds to hla ribosomal binding site and promotes α-hemolysin (hla) translation in S. aureus [57][71]. Similarly, RNAIII differentially regulates the expression of several virulent genes, including hemolysins, repressor of toxins (Rot), toxic shock syndrome toxins (TSST), and several others, as listed in Figure 2.
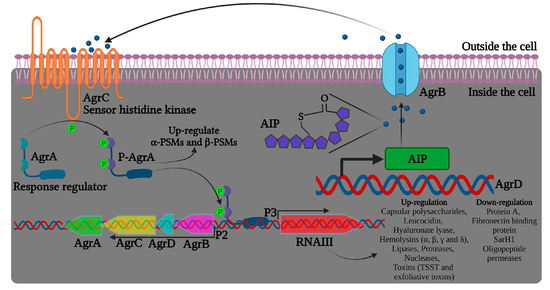
Figure 2. Illustration of the agr quorum sensing system in Staphylococcus aureus and RNAIII regulation of virulence factors. The precursor AIP is encoded by the agrD gene. AgrB is a transmembrane protein likely to be involved in the maturation and transport of the precursor AIP. AIP can bind to AgrC that, together with AgrA, form a TCS signaling system and increase agr activity.
Apart from the agr system, there are other TCS with important roles associated to S. aureus virulence (Table 2) and are considered appealing targets for developing antibacterial drugs [56][70].
In addition to the autoregulatory nature of AgrA, P2 and P3 promoters can be regulated by other players, such as SarA/SarR (staphylococcal accessory regulator), with SarA able to activate and SarR repressing P2 transcription [58][59][72,73]. It was also reported that the TCS srrAB and the global regulator CodY manipulate the agr system expression [60][74]. Moreover, the agr QS system was described to mediate biofilm dispersion by upregulating the extracellular protease in conditions of appreciable biofilm population and low nutritional level [61][62][75,76].
Table 2. Summary of the role of two-component systems and other regulatory genes involved in the virulence genes expression in Staphylococcus aureus [63][64][65][66][67][68][69][77,78,79,80,81,82,83].
| Regulators | Functions |
|---|---|
| AgrCA | Cell-to-cell communication system, where the bacteria communicate with self-produced autoinducing peptide Essential for biofilm disassembly and initial attachment Agr regulation represses adhesins and stimulates phenol-soluble modulins and proteases |
| AirSR/YhcSR | Involved in cellular homeostasis and energy production Important for aerobic and anaerobic growth Positively regulates the expression of the sspABC operon |
| ArlRS | Positive regulator of MgrA and Spx Regulates many cellular processes, including cell wall-anchored adhesins, virulence factors, polysaccharides, and capsular synthesis genes |
| BceRS | Positive regulator of bceAB and vraDE genes Confers resistance towards bacteriocins by transporting bacteriocins outside the cytoplasm through BceAB and VraDE proteins |
| BraSR | Confers resistance towards nisin A and nukacin ISK-1 Exhibits significant regulatory effects on the symbiosis of S. aureus and the type I bacteriocin strain |
| CodY | Strain-dependent regulation of PIA Positive regulator of biofilm formation through the induction of ica operon Cytoplasmic regulator for metabolic response Positive regulator of virulence factor protease |
| GraRS/ApsRS | Belongs to the intramembrane-sensing histidine kinase (IM-HK) familyPositively regulates expression of the dlt operon Essential in evading host defense mechanisms such as neutrophil killing and cationic AMPs |
| HptRS | Hexose phosphate transporter Primarily involved with hptA (initiates autophosphorylation of hptS based on the phosphate concentration), uhpT (downstream regulatory protein transports phosphate/fosfomycin into the bacterial cell to maintain physiological metabolism) Mutation reduces the uptake of fosfomycin (structure similar to phosphoenolpyruvate) and increases the bacterial resistance |
| HssRS | Heme sensor system responding to heme exposure Positive regulator of the efflux pump HrtAB (heme-regulated transporter), which plays a role in maintaining intracellular heme homeostasis Found to have a role in regulating virulence and modulating host immune response |
| KdpDE | Involved in sensing potassium (K+) limitation or salt stress It plays a role in the expression of genes involved in capsule biosynthesis, amino acid and central metabolism |
| LytRS | Regulates cell lysis and induces the expression of irgAB Plays a predominant role in eDNA-mediated biofilm formation |
| MgrA | SarA family cytoplasmic regulator and prime effector of the ArlRS system Repression of adhesins and negative regulator of biofilm formation |
| NreCB | Involved in oxygen sensing; converts nitrate and nitrite as final oxygen acceptors Regulates the gene clusters involved in nitrate (narGHJI) and nitrite reduction (nirRBD) Expression is controlled by NreA, which inhibits NreB autophosphorylation |
| NsaRS | Belongs to the IM-HK family Important in conferring resistance towards nisin |
| PhoRP | Involved in response to inorganic phosphate starvation Positive regulator of pstSCAB and nptA, and also modulates the expression of pitA |
| Rot | SarA family cytoplasmic regulator and prime effector of the Agr QS system Positive regulator of biofilm formation through protease repression and adhesin induction |
| SaeRS | Stimulates adhesin and nuclease production and is very much crucial in the biofilm maturation process |
| SarA | Positive regulator of biofilm formation through the induction of ica operon Induces biofilm formation through the repression of protease production |
| SigB | Strain-dependent regulation of PIA Positive regulator of virulence factors protease and nucleaseEssential for initial attachment and biofilm disassembly Cytoplasmic regulator for stress response |
| SrrAB | Global regulator for S. aureus virulence Critical for survival under environmental stress Regulation of genes involved in anaerobic metabolism, nitrous oxide detoxification, cytochrome biosynthesis and assembly, biofilm formation, hydrogen peroxide metabolism, and programmed cell death |
| VraRS | Associated with vancomycin resistance Essential for cell wall synthesis Positive regulator of PBP2, SgtB, and MurZ |
| WalKR/YycGF | Essential for cell wall metabolism and cell viability Positive regulator of AtlA, SsaA, IsaA, and LytM Regulates the expression of host matrix interaction proteins, cytolytic toxins, and proteins involved in host immune evasion |
6.2. Intercellular Adhesin-Mediated Biofilm Formation and Beyond
Although several factors were reported to influence the attachment and subsequent biofilm formation, the production of extracellular polysaccharide adhesin termed polysaccharide intercellular adhesin (PIA) or polymeric-N-acetylglucosamine (polyGlcNAc) is the best studied biofilm mechanism in Staphylococci [70][84]. PIA is produced by proteins encoded by the intercellular adhesin (ica) gene cassette, including icaR and icaADBC (both regulatory and biosynthesis domains). During biofilm formation, the regulator of biofilm formation, Rbf protein, facilitates the expression of the icaADBC operon by suppressing icaR expression [71][85]. In addition, the ica gene cassette is essential for adhesion to surfaces, like medical devices, by mediating the deacetylation of PIA to produce more positively charged polyGlcNAc molecules [72][86] (Figure 1). However, biofilm development can also be enhanced in an alternative mode (PIA-independent synthesis), even in the absence of the icaADBC operon, especially in ica mutant S. aureus [70][84]. In such ica mutant strains of S. aureus, biofilm production is promoted by Staphylococcus surface protein (SasG) and other biofilm-related proteins, like SasC and protein A (SpA) or accumulation-associated protein or biofilm-associated protein. The extracellular matrix-binding protein (Embp) is another significant player underlying Staphylococcal biofilm formation [73][87]. SasG is a member of the G5-E repeat family, containing G5-E domains and A domain. The N-terminal region of the A domain is linked to cell wall-spanning and anchoring domains. Sequential cleavage of the A domain leads to the exposure of G5-E repeats in the cell membrane, which promote biofilm formation by the adhesion of the G5-E domain of neighboring cells [73][74][87,88].
LuxS is another notable QS system in S. aureus and also in other Gram-positive and Gram-negative bacteria. It has been reported that the autoinducer-2 (AI-2) molecule produced by the luxS gene in S. aureus controls the transcription of the ica operon by repressing the expression of the icaR gene [75][89]. In addition to the PIA synthesis, cellular aggregation in S. aureus has also been mediated by FnBPs (fibronectin-binding proteins) with the aid of Atl (autolysin) and sigB regulation [76][90]. SigB is a positive regulator of biofilm formation by enhancing the synthesis of Clf, FnbpA, and coagulase and repressing the proteins involved in biofilm dispersion [77][91].
7. Role of Mobile Genetic Elements in Antibiotic Resistance and Pathogenesis in Staphylococcus aureus
Mobile genetic elements (MGE) or transposons are responsible for carrying the genes that facilitate resistance to antibiotics, such as methicillin or other β-lactams, in MRSA. The staphylococcal chromosome cassette mec (SCCmec) is one such MGE in S. aureus [78][92]. The SCCmec gene cassette comprises a functional domain (mecA, methicillin resistance determinant), regulatory domain (mecR1 and mecI representing promoter and repressor, respectively), insertion sequence, and ccr (cassette chromosome recombinase gene complex) domain. The ccr domain is responsible for mobilizing the transposon (while integrating into S. aureus DNA) using its recombinase activity. With the att (attachment) site, the integration of the MGE with the bacterial genome is established at a specific site near the origin of replication, OrfX [79][93]. Malachowa and DeLeo [80][94] have sorted out several types of MGE in S. aureus via plasmids, transposons, lysogenic phages, pathogenicity islands (SaPI), and the staphylococcal cassette chromosome (SCC). It was reported that the antibiotic resistance element SCCmec also carries a cytolysin gene encoding PSM. The transfer of such MGE with antibiotic and virulence determinants confers increased virulence, even in the strains that lack genome-encoded PSM production [81][95]. Similarly, the presence of VanA in plasmid confers resistance towards vancomycin in S. aureus, and is believed to be acquired from transposon Tn1546, obtained initially from vancomycin-resistant Enterococcus [82][96].
The genetic analysis of S. aureus variant strains from a broadly infected population showed a correlation containing similar and related markers, such as the SCCmec domain and the Panton-Valentine leukocidin (pvl) locus [22][83][37,97]. This investigation established the relevance of the pvl gene in the formation of abscesses and furuncles [22][37]. The virulence of MRSA associated with community infections is owing to the expression of certain factors like PVL toxin, alpha toxin, and PSM protein (responsible for the degradation of host neutrophils and WBCs) [84][98]. PVL is known as a pore-forming protein in the leucocyte membrane, leading to tissue necrosis [85][99]. Nevertheless, the expression of toxins can be regulated by the agr operon and other regulatory genes [86][100].
Given the difficulties raised by the virulence factors mentioned above and the emergence of MDR strains of S. aureus, the strategies to control and eradicate its infections have become progressively more sophisticated. Hopefully, a combination of rationally used antimicrobial compounds and emerging nanotechnology-enabled approaches will improve the therapeutic paradigm to control S. aureus pathogenesis and infections.