Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mirela Vaduva and Version 2 by Rita Xu.
The development of sensorial platforms based on graphene derivatives and conducting polymers (CPs), alternatively deposited or co-deposited on the working electrode (usually a glassy carbon electrode; GCE) using a simple potentiostatic method (often cyclic voltammetry; CV), possibly followed by the deposition of metallic nanoparticles (NPs) on the electrode surface (ES). These materials have been successfully used to detect an extended range of biomolecules of clinical interest, such as uric acid (UA), dopamine (DA), ascorbic acid (AA), adenine, guanine, and others.
- graphene oxide
- reduced graphene oxide
- conducting polymers
- non-covalent functionalization
- uric acid detection
- sensor
- composites
- polyaniline
- poly(3,4-ethylenedioxytiophene)
- polypyrrole
1. Introduction
Uric acid (UA) is an important biomolecule in the human body, due to its connection with the appearance of certain diseases: an increased level of UA is the major cause for the onset of illnesses, such as gout, hyperuricemia, Lesch–Nyhan syndrome [1], diabetes, kidney, and heart disorders, which can irreversibly affect human individuals. UA, as the chemical compound 7,9-dihydro-1 H-purine-2, 6, 8-(3H)-trione, is the main product of purine metabolism. All mammals, except for the primates, are uricolytic organisms, having the capacity for enzymatic oxidation of UA to allantoin, due to the presence of the enzyme urate oxidase. Significant variations in the UA level are due to an increased catabolic rate or as a consequence of a dysfunction in the elimination route, leading either to increased production of UA or an accumulation of it in different parts of the body. Diagnostic confirmation is achieved through monitoring of UA levels in plasma, urine, and saliva samples.
As UA coexists with many other biomolecules inside the human body, such as AA and DA, the main problem regarding its detection arises from the latter’s interference with UA, whose concentration exceeds the concentration level of DA by a hundred times [2].
Composites based on CPs and carbon NPs (CNPs) [5] decorated with metallic oxides [4] and/or metallic NPs [6] have been successfully used for the selective and sensitive detection of an extended range of biomolecules of clinic interest, such as UA, DA, AA, adenine, guanine, epinephrine, norepinephrine, and so on.
Due to their special properties, nanocomposites can significantly improve the working parameters of sensors, when compared to the individual components [7].
In t
Thise work, researchers focus theirmain attention on composites based on CPs of the Polypyrrole (PPY), poly(3, 4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI), and polyimidazole (PIm) type, as well as CNPs of the graphene oxide (GO), reduced graphene oxide (RGO), and graphene quantum dots (GQDs) type.
Each of the main components within the composite material has a major contribution to sensor performance enhancement; more precisely, regarding the improvement of the electrocatalytic property as well as the sensitivity and selectivity of the electrochemical response. For example, CPs provide the advantage of increased active surface area, in some cases ensuring a porous structure of the electrode, as is the case for PPy [5], which enhances the stability and adhesion properties of the electrode surface. Due to the abovementioned properties, the active specific area facilitates electronic transfer and improves the adsorption of the target bio-analytes at the ES by promoting electro-oxidation of the adsorbed molecules. Monomers that readily polymerize on the ES, assisted by a dopant which helps to increase conductivity, leads to CPs with the enhanced catalytic activity. The class of CPs includes many representatives with very good conductors that help increase conductivity, such as PPy [5][6][5,6], PEDOT [8], PIm [9], and others [10][11][10,11].
PPy, which has a conjugated structure, is one from the most important CPs and has been successfully used in the electro-detection of various biomolecules [12][13][14][12,13,14]. This is due to several of its properties, including high conductivity, ease of preparation, and good stability. Furthermore, PPy can be electrochemically oxidated to OPPy (over-oxidated PPy), incorporating a high number of carbonyl groups after the oxidation process [15], thus providing unique cationic selectivity, excluding the anionic species to reach the level of ES [12][16][12,16] selectivity which is desirable for the detection of biomolecules including DA and UA, among others. Over-oxidation of PPy can lead to a disruption of conjugation and, as such, although OPPy is a good ionic conductor with ion-selective properties, it may also present non-conductive electronic properties [13][16][13,16], which could diminish the electrochemical signal collection.
Overall, despite its ease of synthesis, good catalytic activity, low cost, good polymerization yield, and significant chemical, thermal, and mechanical stability, together with good adhesion and high surface area, PPy has also important drawbacks, including high charge transfer resistance and low conductivity [17], which can easily be addressed by combination with conductive carbon structures (i.e., incorporation into hybrid structures).
Another CP often used for electro-detection of UA is PANI. This polymer provides a good matrix for different inorganic semiconductors and carbonic materials, due to its high relative conductivity, chemical and electrochemical stability, and other advantages such as ease of preparation, reduced cost of monomers and economic process of fabrication, reversible redox behavior, and thermal stability. The most active form of PANI has been reported to be emeraldine, due to its high active area and low charge transfer resistance (RCT) [17]. However, one important problem remains; namely, sufficient conductivity for applications in sensors field, making it suitable for combination with graphene derivatives or other highly conductive structure which, on their own, cannot allow for a uniform layer deposition on the ES with enhanced selectivity and, further, with extended electro-catalytic activity.
The study conducted by Ghanbari, K. et al., supported by that of Pihel, K. et al. [4][13][4,13], has revealed that, by modifying the working ES with composite material based on PANI and graphene, the performance of the sensor could be improved. This fact was translated through an increase in the electronic transfer rate as a consequence of the strong electronic interaction which takes place between graphene and PANI.
PEDOT is another of the most-studied nanostructured polymers, due to its high electric conductivity [18], transparency [19], and high-quality film preparation [20]. As has been reported by many research groups, including that of Reddy, S. et al., PEDOT can be successfully used as an electrochemical sensor in numerous applications, mainly due to its capacity to increase the electrode’s specific area [21][22][21,22].
Still, PEDOT alone, with its high conductivity at room temperature, electrochemical reversible behavior, and important thermal and chemical stability, has poor cycling stability and low capacity to form charge transfer channels within its structure, providing low catalytic activity. [23]
Besides these conductive polymers, PIm has been also reported, as pristine or in combination with carbo[1]n nanostructures, and tested for UA detection. Polymerized PIm creates a uniform layer on the ES, which is chemically stable and has controllable thickness [24], while the over-oxidation process improves its permselectivity and antifouling process. The combination of PIm with highly conductive compounds can improve its electro-catalytic response [9].
Overall, CPs lead to improvements in the sensitivity and stability of sensors while, at the same time, ensuring the good mechanical strength of the final structure of the composite. The combination of high conductivity and biocompatibility, together with the capacity to immobilize biomolecules on the ES for a long period of time while maintaining their full activity, indicate the high potential of CPs for biosensor construction.
On the other hand, carbon nanostructures present controllable electronic, hydrophilicity (e.g., GO and GQDs), stability, and catalytic properties, due to the functional groups on their surface, as well as high biocompatibility and large surface areas, providing mechanical strength and high conductivity to the composite structure [25].
From the category of graphene derivatives used in the electro-detection of UA, GO is often used, due to its active surface area, water-dispersion capacity and hydrophilicity, thermal and mechanical properties, versatility, and ease of surface modification. A major disadvantage of GO is the tendency of stacking, thus restoring the graphite structure; this disadvantage can be overcome, for instance, with the help of CPs [5][6][5,6].
Various functional groups rich in oxygen at the GO surface allow for good dispersion of GO in aqueous solution; however, its conductivity may be lost due to the large discontinuity in the sp2 hybrid carbon network. This is another reason why the combination of GO and CPs has high potential, particularly when referring to materials designed for electrode modification in order to achieve better performance in electrochemical detection.
Another graphene derivative with promising applications in electro-detection for a wide range of analytes is RGO, which possesses good properties for sensors, either by itself [26] or as a component of composite materials from the working ES [4][27][28][29][4,27,28,29]. Due to its high conductivity and significant charge mobility, RGO allows for an increased sensitivity of detection—one of the most fundamental properties of a sensor—by accelerating the CT rate together with a high specific surface area [26]. Another advantage of using RGO is the high number of active sites, as a consequence of the numerous functional groups on its surface, contributing to a uniform deposition of the conductive polymer. Further, the π–π stacking interaction between the CPs and the RGO layer contributes to an enhanced composite conductivity, facilitating charge transport and determining shorter routes for ion diffusion. A significant shortcoming of RGO is its tendency to stack sheets together, leading to a smaller active surface area, which can be overcome by mixing with CPs, which play a stabilizing role in hindering RGO layer aggregation and provide better performance of the working electrode on which the composite material has been deposited.
In the study conducted by Yola, M.L. et al., it has been reported that GQDs, a form of graphene derivatives, when used as part of the modified electrodes used for UA detection, very good response was achieved [30]. GQDs correspond to zero-dimensional (OD) structures with diameter smaller than 10 nm, exhibiting quantum confinement effects and significant edge effects. [31] The main properties of GQDs are (a) high solubility in H2O; (b) low toxicity; (c) bio-compatibility; (d) photoluminescence in visible range; and (e) tunable bandgap. All of these properties recommend them for use in various applications, such as bio-imaging [32][33][32,33].
Further, composites based on CPs and graphene derivatives such as GO, RGO, and GQDs have been synthesized mainly using three methods: (i) mixing of the constituents, including mechanical mixture [34] or electromagnetic stirring of the mixing solution [35]; and (ii) chemical or (iii) electrochemical polymerization of the monomer in the presence of graphene derivatives [36][37][38][39][36,37,38,39]. The prevalent method is electrochemical synthesis [40]. Composites can be prepared through the simultaneous [41] or alternative [4] deposition of CPs and graphene derivatives. The major advantage of using this method is the good control of the film thickness, achieved by varying certain parameters such as work potential, current intensity, scan speed, deposition time, and number of cyclic voltammograms. Furthermore, the deposition process is carried out directly on the ES, using small amounts of analytes, with minimal losses.
The electrochemical method can also be used for UA determination, due to important advantages such as rapid detection, low preparation costs, reduced amounts of production materials, and ease of handling, together with the sensitivity and selectivity of the modified electrode [42]. Such electrochemical detection is usually carried out on flat electrodes made from carbon-based materials, such as glassy carbon [5][43][5,43], carbon fiber paper [6], carbon paste electrode [44], or ionic liquid-modified screen-printed carbon [45].
In order to increase the sensitivity, the ES is sometimes modified with metallic NPs, carbon nanotubes, carbon monoliths, quantum dots, graphene and its derivatives, and/or CPs [2]. According to the studies reported in the last 6 years, the most promising results for achieving the highest sensitive electrochemical signal on a modified electrode, with linear response in a large range of concentrations and the lowest limit of detection (LOD), turned out to be composites based on carbonic structures and CPs, decorated with metallic NPs [1][3][4][1,3,4].
2. Chemical and Vibrational Properties of the CPs/GO Composites
2.1. Chemical and Vibrational Properties of GO/PIm Composites
The main advantage of using GO/PIm composites is their fast preparation, which can be performed directly on the ES. They can be synthesized through electrochemical co-deposition from a suspension of GO and imidazole (Im). After deposition, the composite can be easily electrochemically over-oxidized. During this process, multiple functional groups rich in oxygen are introduced into the imidazole unit, thus improving the permselectivity and sensor response [9]. A GO/PIm composite has been prepared, according to the electrochemical method described by Liu, X. et al., right on the GCE ES, from 0.3 moles of imidazole dispersed in a mixture of 3 mL GO suspension (5 mg/mL) and 0.3 moles of surfactant (sodium dodecyl sulfate; SDS), with a potential range between −0.2 and 0.8 V. The composite material, PIm–GO, was then oxidized in 0.1 M phosphate-buffered saline (PBS) medium (pH = 3) at +1.8 V for 250 s [9]. When analyzed by FTIR spectroscopy, the PImox–GO composite presented IR bands characteristic of both PIm and GO. Therefore, the IR bands assigned to the C=C vibrational mode of the aromatic ring and the C=N– stretching vibration, located at 1642 and 1468 cm−1, respectively, were attributed to PIm, together with the bands situated at 2850 and 2918 cm−1 belonging to the stretching vibration of C–H bonds [46][47][46,47]. In addition, the corresponding C=C bond band (1642 cm−1) in the PIm–GO spectrum was stronger than that for PIm. This, together with the new bands that emerged at 1204 and 3435 cm−1, attributed to the C–O and O–H bonds of GO, respectively [48], demonstrated the formation of the nanocomposite.2.2. Chemical and Vibrational Properties of GO/PPy Composites
To improve its electro-conductivity, GO has been combined with PPy, in this way promoting the electrochemical response of the modified sensor due to the π–π stacking between GO layers and PPy rings [6]. The nanocomposites were co-deposited on carbon fiber paper (CFP) using the potentiostatic method, followed by the deposition of AuNPs on the composite surface through CV. The co-deposition was performed using a mix of GO suspension (1 mg/mL), pyrrole monomer (50 mM), Na2SO4 0.1 M, and 20 mM SDS, ultrasonicated for 30 min at constant potential of +0.7 vs. Ag/AgCl, using a three-electrode configuration consisting of Ag/AgCl as the reference electrode, Pt wire as the counter electrode, and CFP as the working electrode. The non-covalent functionalization pattern is also present for the GO composite with PPy (see Figure 1); in this case, the Py monomers are attached onto the GO surface through π–π interactions, hydrogen bonds, and VW forces [49]. After attaching to the GO surface, the pyrrole monomers were electrochemically oxidated, without stacking as a multiple layer structure. The structure of GO deposited on the electrode resembled wrinkled paper with a high active area, while PPy deposited on the top of GO could not be distinguished due to its dispersion on the GO surface, rather than stacking and overlapping as multiple layers. This was considered to occur due to the strong interaction between the GO platform and Py dissociative monomers. In contrast, the pristine PPy deposited on CFP appeared different to the composite, presenting an agglomerated structure.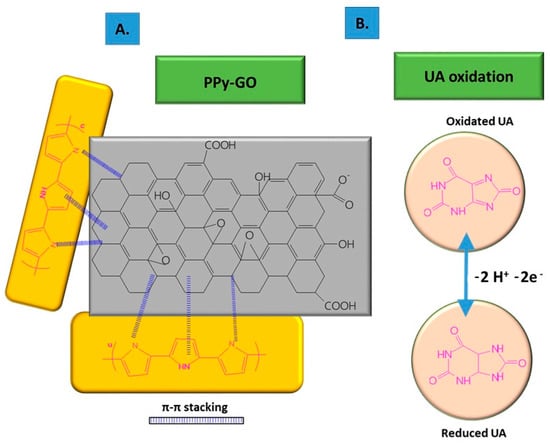
Figure 1. Non-covalent functionalization of GO with PPy inside the composite (A); and UA oxidation process at the surface of the composite deposited onto ES (B).
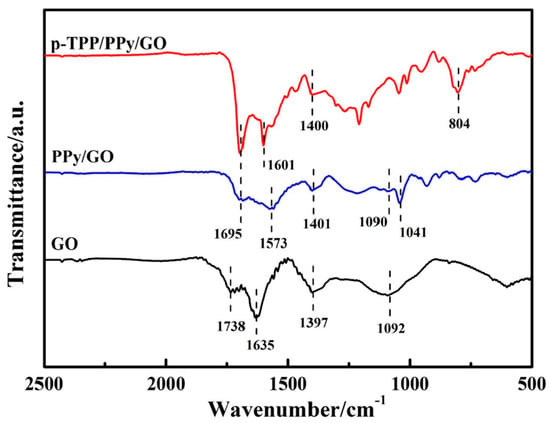
Figure 2. FTIR spectra of the compounds: GO (black curve), PPY/GO (blue curve), and p-TPP/PPY/GO (red curve). Reproduced with permission from ref. [5]; Copyright 2022 Springer.
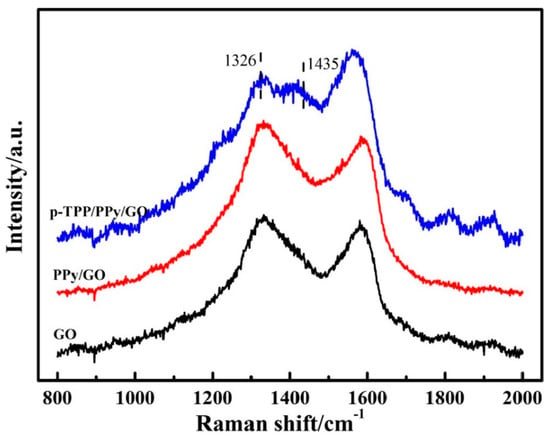
Figure 3. Raman spectra of the compounds: GO (black curve), PPY/GO (red curve), and p-TPP/PPY/GO (blue curve). Reproduced with the permission from ref. [5]; Copyright 2022 Springer.
2.3. Chemical and Vibrational Properties of GO/PEDOT Composites
Furthermore, GO can play the role of a dopant in certain composites, balancing the charges; for example, in the case of PEDOT/GO, it contributes to balancing the positive charges from the PEDOT backbone by increasing the amount of functional groups with oxygen [8][59][60][8,59,60]. In order to improve the GO conductivity, which is weakened by the sp2 interrupted network of C, new composites with PEDOT were tested, and the synergic effect of both PEDOT and GO led to improved electrocatalytic properties of the resulting sensor [8]. A small amount of GO suspension (1 mg/mL) in deionized water mixed with 3,4-ethylenedioxythiophene (EDOT) monomer (10 mM) was deposited on ITO and covered with Whatman paper, in order to obtain a thin layer electrochemical cell. The piece of paper retains not only the reactants, but also connects the electrode system. The electrochemical polymerization was carried out using chronoamperometry, by applying a potential of 1.2 V for a period of 150 s. At the end, a PEDOT–GO/ITO sensor was obtained [8]. The positive charge of the resulting polymer can attract negatively charged groups from the GO surface and, so, the interaction taking place between the two components is described as non-covalent functionalization [8] (see Figure 4 below). During the polymerization process of the PEDOT–GO nanocomposite on ITO substrate, the composite benefits from the mechanical support of the GO layer, which provides increased stability. Analysis of the composite surface morphology revealed that the PEDOT–GO film was uniformly distributed in a random form, appearing as a porous and rough network. The free space between the layers/sheets is favorable for electron exchange between the biomolecules and the electrode substrate. Overall, combining the components PEDOT and GO resulted in an increase in the sensitivity of the sensor [8].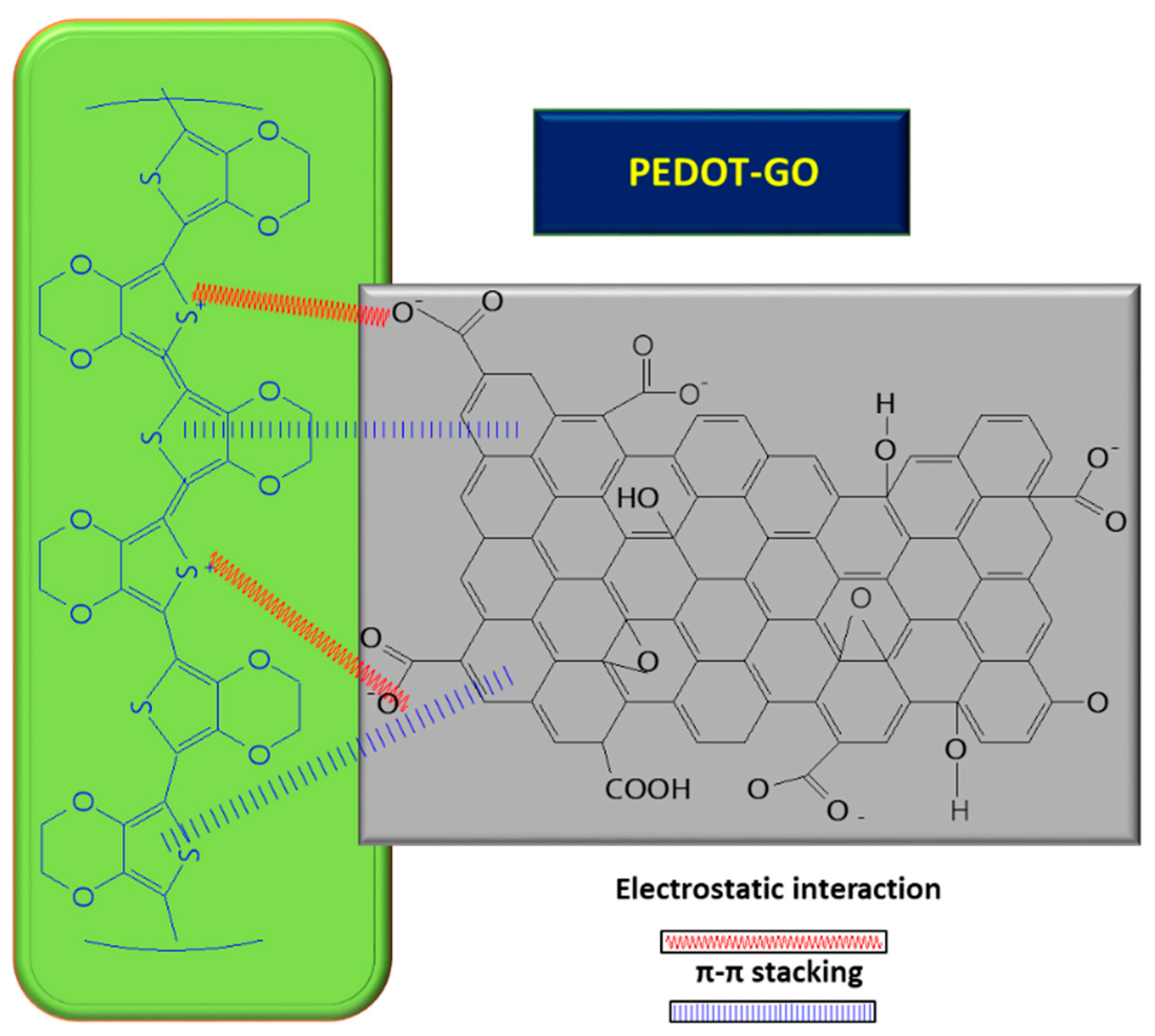
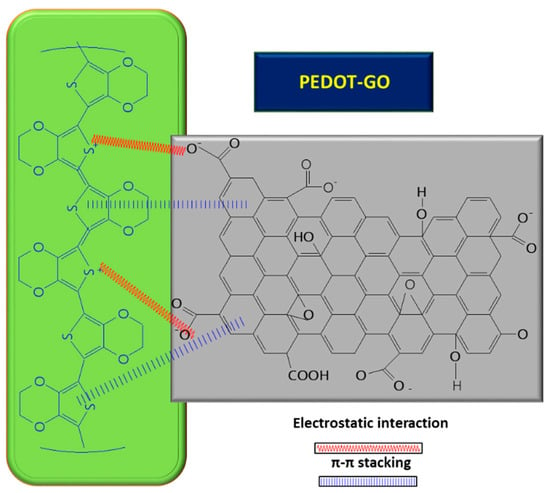
Figure 4. Interaction between GO and PEDOT, inside the composite used for UA detection.
3. Chemical and Vibrational Properties of CPs/RGO
The methods of synthesis used to obtain this type of composite range from the simplest and fastest, such as electrochemical methods [4][63][4,70], to complex routes for preparation of composites; for example, through a hydrothermal route [27]. Among the CPs, the most-reported in combination with RGO to obtain composites used for UA detection are PPy [64][71], PANI [4][27][4,27], and PEDOT [65][72].
3.1. Synthesis and Interaction between RGO and PPy Inside the Composite
According to the study published by Chen, X. et al. [64][71], composites based on PPy and RGO were prepared starting with the deposition of RGO film on top of the GCE. Next, using a Py solution (0.2 M) in PBS medium (10 mM, pH = 7.4), within the potential range of −0.2 to 0.8 V, the PPy film was deposited on the RGO/GCE surface. The final stage involved PPy film oxidation, performed through an electrochemical process within the potential range of 0–1 V, during 4 cyclic voltammograms in NaOH 0.1 M solution. The electrochemical properties of GCE modified with OPPy (over-oxidated PPy) and RGO were tested using redox samples, including negatively charged Fe (CN)63− and positively charged Ru (NH3)63+, comprising electroactive compounds of similar size. On PPy/ERGO/GCE, the charge currents increased and the redox maxima disappeared. On the other hand, when using OPPy, within OPPy/ERGO/GCE, the charge currents decreased, the ruthenium redox maxima disappeared at an oxidation potential of 0.253 V, and a reduction potential of −0.237 V was observed. The oxidation maximum increased by 4.7 times and the reduction potential by 9.3 times, compared to GCE alone. According to [64][71], these results indicated that the ruthenium sample reached the ES and CT took place. The hybrid nanocomposite OPPy/ERGO has ion-selective properties, as the negative charges of OPPy allow only cations to reach the ES and be involved in the CT process. Comparing the electrochemical behavior with both the positively and negatively charged systems, the influence of the PPy over-oxidation process on the electrochemical activity of PPy/ERGO hybrid composite was highlighted. The negatively charged OPPy film led to electrostatic adsorption. On the PPy/RGO film surface, with the help of SEM images, it was observed that small PPy spheres covered the entire surface. This indicated the formation of a 3D polymeric structure through in situ polymerization had occurred on the ES, with the help of a graphene template [66][73]. After performing the over-oxidation treatment on the ES, a rough, compact, and uniform film was obtained, resembling sheets of wrinkled paper with many edges.
Another method for synthesis of an RGO/PPy composite involved co-synthesis from a mixture of GO (40 mg) and cetrimonium bromide (91 mg), phosphoric acid (15 mL), and Py monomer, which was gradually added to the initial mixture and stirred for about 2 h at 15 °C [29]. Polymerization was initiated by adding ammonium persulfate solution (APS). After 4 h of reaction, the black precipitate was filtered, washed, and dried. RGO/Pd@PPy nanocomposite was obtained by dispersion of RGO/PPy powder in ethylene glycol and 7 mg/mL PdCl2 solution. Then, the mixture was exposed to microwaves for thermal treatment for 2 min, following which it was centrifuged and dried at 100 °C under vacuum. The RGO/Pd@PPy NPs was then drop-casted on the GCE surface. According to the Raman spectroscopy analysis, the ID/IG ratios reported for RGO/PPy and RGO/Pd@PPy were equal to 0.87 and 0.96, respectively, which was interpreted as proof of the functionalization of RGO with PPy–Pd [29]. Comparing the SEM images recorded on PPy/ERGO before and after the over-oxidation process, it was found that the PPy/ERGO film had a laminated structure before oxidation, with multiple spheres of PPy covering the PPy/ERGO/GCE surface, suggesting the generation of a 3D polymeric structure on the ES. According to the results obtained by Demirkan, B., the ERGO layer, deposited before starting the polymerization of Py, plays the role of a template, guiding the entire process of the PPy film growth. At the end of the deposition process, after the over-oxidation stage, a compact, uniform, and thin hybrid film of PPy/ERGO was formed [29].
3.2. Chemical and Vibrational Properties of RGO/PANI Composites
A PANI/RGO composite has been prepared through chemical synthesis from GO, hydrothermally treated with a mixture containing Fe(NO3)3, SnSO4, and PANI, obtained through oxidative chemical polymerization from 500 mg ANI, 5 mg surfactant (Triton X-100), 20 mL ultrapure water, and 10 mL HCl solution (1 M) [27]. According to the synthesis reported by Minta, D. et al., a mixture of ANI and surfactants, in an acidic medium, was ultrasonicated for 30 min at a temperature between 0–5 degrees, after which polymerization was initiated through the addition of ammonium persulfate. The final composite was synthesized by adding 50 mg of GO decorated with tin and iron oxides in ultrapure water and ultrasonicated during 30 min. Then, PANI powder was added, and the reaction took place in an autoclave at 180 °C, for 12 h, under stirring. After several wash/dry cycles, the resulting composite had a mass ratio 50:17:33 PANI:Fe2O3–SnO2:RGO (PFSG).
The PANI/RGO formed through in situ electrochemical deposition presented a mixed morphology, having both the RGO layer structure and the PANI nanofibers distributed on the graphene layer surface, obtained after performing the ANI polymerization in HCl [4]. The main IR bands from the PANI spectrum, located at 1390, 3445, and 2830 cm−1, were assigned to the stretching vibrational mode of C–N, N–H, and C–H bonds, respectively. To these, the stretching vibrational mode of quinoid ring, located at 1138 cm−1 (N–Q–N–Q) was added. The prominent absorption bands were assigned to C=C deformation from the quinoid ring (1585, 1498 cm−1) and to the stretching vibrational mode of the C–N bond of the secondary aromatic amine [67][63]. In the FTIR spectra of RGO/PANI, the absorption bands from 1560 and 1487 cm−1 were assigned to the stretching vibrational mode of C=C bonds within quinoid and benzene structures, respectively [68][74]. The bands from 1296 and 1242 cm−1 were attributed to the stretching vibrational mode for C–N and C=N bonds, respectively [69][75].
These bonds suggest the coverage of RGO sheets with PANI, and the results provide evidence of the functionalization type which takes place between PANI and RGO; namely, covalent functionalization [70][76] (see Figure 5).
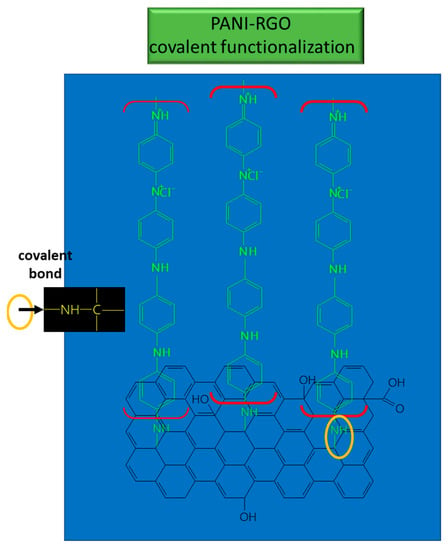
Figure 5. Interaction between RGO and PANI in the composite used for UA detection.
The presence of these bands suggest the coverage of ZnO with PANI/RGO, resulting in a ZnO/PANI/RGO composite, which leads to a shift in the position of peaks and changes in their relative intensity. Regarding the ZnO/PANI/RGO FTIR spectrum, the peaks located at 1560, 1487, and 1390 cm−1 shifted towards the higher wavenumbers of 1566, 1488.9, and 1394 cm−1, respectively, indicating that the nitrogen atoms from amine groups (NH) and imine groups (N) had bonded with Zn2+ through protonation and complexation reactions [71][77]. The band of high intensity from 1394 cm−1 and the wide band from 437 cm−1 correspond to the stretching vibration of O–Zn–O [72][78]. The strong stretching vibrations of C–N+ bonds, represented by the Raman line at 1330 cm−1, correspond to radical cationic structures, such as the protonated semi-quinoid structure. The lines situated at 1225 cm−1 and 1170 cm−1 could be assigned to the stretching vibration of C–N bonds in the polaronic units and in-plane bending vibrations of C–H bonds corresponding to bipolaronic shapes, respectively. The Raman lines from 829, 770, 507, and 411 cm−1, correspond to the bending vibration of the C–N–C bond, to the quinoid ring deformation, and to in-plane and out-of-plane amine deformation, respectively. The Raman lines from 1485 and 1583 cm−1 were assigned to the RGO signature shift from the initial position, meaning that a bond between C–N+ and carboxylate group from the RGO sheets was created, which was formed on the basis of the π–π* interactions occurring between PANI and RGO [73][79]. Confirmation of the incorporation of RGO into the PANI matrix during electro-polymerization was recorded through the two bands at 2770 and 3070 cm−1, which were assigned to the respective contributions of the D and G bands, together with the contribution of the 2 D band from the Raman spectra of PANI/RGO composite [74][75][80,81].
References
- Shibasaki, K.; Kimura, M.; Ikarashi, R.; Yamaguchi, A.; Watanabe, T. Uric acid concentration in saliva and its changes with the patients receiving treatment for hyperuricemia. Metabolomics 2012, 8, 484–491. Yola, M.L.; Atar, N. Functionalized graphene quantum dots with bi-metallic nanoparticles composite: Sensor application for simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan. J. Electrochem. Soc. 2016, 163, B718–B725, 2.1191614jes.
- Elangovan, A.; Sudha, K.; Jeevika, A.; Bhuvaneshwari, C.; Kalimuthu, P.; Balakumar, V. Construction of ternary coupled with poly-L_ethionine nanocomposite as a robust platform for electrochemical recognition of uric acid in diabetic patients. Colloid Surf. A 2020, 602, 125050. Zhang, F.; Gu, S.; Ding, Y.; Zhang, Z.; Li, L. A novel sensor based on electropolymerization of beta-cyclodextrin and L-arginine on carbon paste electrode for determination of fluoroquinolones. Anal. Chim. Acta 2013, 770, 53–61, j.aca.2013.01.052.
- Kucherenko, I.; Soldatkin, O.; Kucherenko, D.; Soldatkina, O.; Dzyadevych, S. Advances in nanomaterial application in enzyme-based electrochemical biosensors: A review. Nanoscale Adv. 2019, 1, 4560–4577. Yola, M.L.; Atar, N. Functionalized graphene quantum dots with bi-metallic nanoparticles composite: Sensor application for simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan. J. Electrochem. Soc. 2016, 163, B718–B725, 2.1191614jes.
- Ghanbari, K.; Moludi, M. Flower-like ZnO decorated polyaniline/reduced graphene oxide nanocomposites for simultaneous determination of dopamine and uric acid. Anal. Biochem. 2016, 512, 91–102. Zhang, D.; Li, L.; Ma, W.; Chen, X.; Zhang, Y. Electrodeposited reduced graphene oxide incorporating polymerization of l-lysine on electrode surface and its application in simultaneous electrochemical determination of ascorbic acid, dopamine and uric acid. Mat. Sci. Eng. C 2017, 70, 241–249, j.msec.2016.08.078.
- Dai, H.; Wang, N.; Wang, D.; Zhang, H.; Ma, H.; Lin, M. Voltammetric uric acid sensor based on a glassy carbon electrode modified with a nanocomposite consisting of polytetraphenylporphyrin, polypyrrole, and graphene oxide. Microchim. Acta 2016, 183, 3053–3059. He, S.; He, P.; Zhang, X.; Zhang, X.; Liu, K.; Jia, L.; Dong, F. Poly(glycine)/graphene oxide modified glassy carbon electrode: Preparation, characterization and simultaneous electrochemical determination of dopamine, uric ac-id, guanine and adenine. Anal. Chim. Acta 2018, 2016, 75–82, j.aca.2018.06.020.
- Tan, C.; Zhao, J.; Sun, P.; Zheng, W.; Cui, G. Gold nanoparticles decorated polypyrrole/graphene oxide nanosheets as a modified electrode for simultaneous determination of ascorbic acid, dopamine and uric acid. New J. Chem. 2020, 44, 4916–4926. Arulraj, A.; Arunkumar, A.; Vijayan, M.; Viswanath, K.; Vasantha, V. A simple route to develop highly porous nanopolypyr role/reduced graphene oxide composite film for selective determination of dopamine. Electrochim. Acta 2016, 206, 77–85, j.electacta.2016.04.134.
- Dakshayini, B.; Reddy, K.; Mishra, A.; Shetti, N.; Malode, S.; Basu, S.; Naveen, S.; Raghu, A. Role of conducting polymer and metal oxide-based hybrids for applications in amperometric sensors and biosensors. Microchem. J. 2019, 147, 7–24. Pihel, K.; Walker, Q.; Wightman, R. Overoxidized polypyrrole-coated carbon fiber microelectrodes for dopamine measurements with fast-scan cyclic voltammetry. Anal. Chem. 1996 , 68, 2084–2089, ac960153y.
- Huang, X.; Shi, W.; Li, J.; Bao, N.; Yu, C.; Gu, H. Determination of salivary uric acid by using poly(3,4-ethylenedioxythiophene) and graphene oxide in a disposable paper-based analytical device. Anal. Chim. Acta 2020, 1103, 75–83. Gao, Y.; Xu, J.; Lu, L.; Wu, L.; Zhang, K.; Nie, T.; Zhu, F.; Wu, Y. Overoxidized polypyrrole/graphene nanocomposite with good electrochemical performance as novel electrode material for the detection of adenine and guanine.. Biosens. Bioelectron. 2014, 62, 261–267, j.bios.2014.06.044.
- Liu, X.; Zhang, L.; Wei, S.; Chen, S.; Ou, X.; Lu, Q. Overoxidized polyimidazole/graphene oxide copolymer modified electrode for the simultaneous determination of ascorbic acid, dopamine, uric acid, guanine and adenine. Biosens. Bioelectron 2014, 57, 232–238. Beck, F.; Braun, P.; Oberst, M. Organic electrochemistry in the solid state-overoxidation of polypyrrole.. Ber Bunsenges. Phys. Chem. 1 1987, 91, 967–974, https://doi.org/10.1002/bbpc.19870910927.
- Zhang, D.; Li, L.; Ma, W.; Chen, X.; Zhang, Y. Electrodeposited reduced graphene oxide incorporating polymerization of l-lysine on electrode surface and its application in simultaneous electrochemical determination of ascorbic acid, dopamine and uric acid. Mat. Sci. Eng. C 2017, 70, 241–249. Tukimin, N.; Abdullah, J.; Sulaiman, Y. Development of a PrGO-modified electrode for uric acid determination in the presence of ascorbic acid by an electrochemical technique. . Sensors 2017, 17, 1539, s17071539.
- He, S.; He, P.; Zhang, X.; Zhang, X.; Liu, K.; Jia, L.; Dong, F. Poly(glycine)/graphene oxide modified glassy carbon electrode: Preparation, characterization and simultaneous electrochemical determination of dopamine, uric ac-id, guanine and adenine. Anal. Chim. Acta 2018, 2016, 75–82. He, S.; He, P.; Zhang, X.; Zhang, X.; Liu, K.; Jia, L.; Dong, F. Poly(glycine)/graphene oxide modified glassy carbon electrode: Preparation, characterization and simultaneous electrochemical determination of dopamine, uric ac-id, guanine and adenine. Anal. Chim. Acta 2018, 2016, 75–82, j.aca.2018.06.020.
- Arulraj, A.; Arunkumar, A.; Vijayan, M.; Viswanath, K.; Vasantha, V. A simple route to develop highly porous nanopolypyrrole/reduced graphene oxide composite film for selective determination of dopamine. Electrochim. Acta 2016, 206, 77–85. Arulraj, A.; Arunkumar, A.; Vijayan, M.; Viswanath, K.; Vasantha, V. A simple route to develop highly porous nanopolypyr role/reduced graphene oxide composite film for selective determination of dopamine. Electrochim. Acta 2016, 206, 77–85, j.electacta.2016.04.134.
- Pihel, K.; Walker, Q.; Wightman, R. Overoxidized polypyrrole-coated carbon fiber microelectrodes for dopamine measurements with fast-scan cyclic voltammetry. Anal. Chem. 1996, 68, 2084–2089. Pihel, K.; Walker, Q.; Wightman, R. Overoxidized polypyrrole-coated carbon fiber microelectrodes for dopamine measurements with fast-scan cyclic voltammetry. Anal. Chem. 1996 , 68, 2084–2089, ac960153y.
- Gao, Y.; Xu, J.; Lu, L.; Wu, L.; Zhang, K.; Nie, T.; Zhu, F.; Wu, Y. Overoxidized polypyrrole/graphene nanocomposite with good electrochemical performance as novel electrode material for the detection of adenine and guanine. Biosens. Bioelectron. 2014, 62, 261–267. Gao, Y.; Xu, J.; Lu, L.; Wu, L.; Zhang, K.; Nie, T.; Zhu, F.; Wu, Y. Overoxidized polypyrrole/graphene nanocomposite with good electrochemical performance as novel electrode material for the detection of adenine and guanine.. Biosens. Bioelectron. 2014, 62, 261–267, j.bios.2014.06.044.
- Beck, F.; Braun, P.; Oberst, M. Organic electrochemistry in the solid state-overoxidation of polypyrrole. Ber Bunsenges. Phys. Chem. 1987, 91, 967–974. Beck, F.; Braun, P.; Oberst, M. Organic electrochemistry in the solid state-overoxidation of polypyrrole.. Ber Bunsenges. Phys. Chem. 1 1987, 91, 967–974, https://doi.org/10.1002/bbpc.19870910927.
- Tukimin, N.; Abdullah, J.; Sulaiman, Y. Development of a PrGO-modified electrode for uric acid determination in the presence of ascorbic acid by an electrochemical technique. Sensors 2017, 17, 1539. Tukimin, N.; Abdullah, J.; Sulaiman, Y. Development of a PrGO-modified electrode for uric acid determination in the presence of ascorbic acid by an electrochemical technique. . Sensors 2017, 17, 1539, s17071539.
- Saranya, K.; Rameez, M.; Subramania, A. Developments in conducting polymer based counter electrodes for dye-sensitized solar cells–an overview. Eur. Polym. J. 2015, 66, 207–227. Saranya, K.; Rameez, M.; Subramania, A. Developments in conducting polymer based counter electrodes for dye-sensitized solar cells–an overview. Eur. Polym. J. 2015, 66, 207–227, j.eurpolymj.2015.01.049.
- Gueye, M.; Carella, A.; Massonnet, N.; Yvenou, E.; Brenet, S.; Faure, V.; Poget, S.; Rieutord, F.; Okuno, H.; Be-nayad, A.; et al. Structure and dopant engineering in PEDOT thin films: Practical tools for a dramatic conductivity enhancement. Chem. Mater. 2016, 28, 3462–3468. Gueye, M.; Carella, A.; Massonnet, N.; Yvenou, E.; Brenet, S.; Faure, V.; Poget, S.; Rieutord, F.; Okuno, H.; Be-nayad, A.; et al.et al. Structure and dopant engineering in PEDOT thin films: Practical tools for a dramatic conductivity enhancement. Chem. Mater. 2016, 28, 3462–3468, acs.chemmater.6b01035.
- Xia, Y.; Sun, K.; Ouyang, J. Solution-processed metallic conducting polymer films as transparent electrode of optoelectronic devices. Adv. Mater. 2012, 24, 2436–2440. Xia, Y.; Sun, K.; Ouyang, J. Solution-processed metallic conducting polymer films as transparent electrode of optoelectronic devices. Adv. Mater. 2012, 24, 2436–2440, adma.201104795.
- Vreeland, R.; Atcherley, C.; Russell, W.; Xie, J.; Lu, D.; Laude, N.; Porreca, F.; Heien, M. Biocompatible PEDOT:Nafion composite electrode coatings for selective detection of neurotransmitters in vivo. Anal. Chem. 2015, 87, 2600–2607. Vreeland, R.; Atcherley, C.; Russell, W.; Xie, J.; Lu, D.; Laude, N.; Porreca, F.; Heien, M. Biocompatible PEDOT:Nafion composite electrode coatings for selective detection of neurotransmitters in vivo. Anal. Chem. 2015, 87, 2600–2607, ac502165f.
- Yang, G.; Kampstra, K.; Abidian, M. High performance conducting polymer nanofiber biosensors for detection of biomolecules. Adv. Mater. 2014, 26, 4954–4960. Yang, G.; Kampstra, K.; Abidian, M. High performance conducting polymer nanofiber biosensors for detection of biomolecules. Adv. Mater. 2014, 26, 4954–4960, adma.201400753.
- Reddy, S.; Xiao, Q.; Liu, H.; Li, C.; Chen, S.; Wang, C.; Chiu, K.; Chen, N.; Tu, Y.; Ramakrishna, S.; et al. Bionanotube/poly(3,4-ethylenedioxythiophene) nanohybrid as an electrode for the neural interface and dopamine sensor. ACS Appl. Mater. Interfaces 2019, 11, 18254–18267. Reddy, S.; Xiao, Q.; Liu, H.; Li, C.; Chen, S.; Wang, C.; Chiu, K.; Chen, N.; Tu, Y.; Ramakrishna, S.; et al.et al Bionanotube/poly(3,4-ethylenedioxythiophene) nanohybrid as an electrode for the neural interface and dopamine sensor. ACS Appl. Mater. Interfaces 2019, 11, 18254–18267, acsami.9b04862.
- Ahmad, M.S.; Hamdan, K.S.; Rahim, N.A. Towards the Conducting Polymer Based Catalysts to Eliminate Pt for Dye Sensitized Solar Cell Applications. In Advances in Hybrid Conducting Polymer Technology; Engineering Materials; Shahabuddin, S., Pandey, A.K., Khalid, M., Jagadish, P., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 311–326. Ahmad, M.S.; Hamdan, K.S.; Rahim, N.A. Towards the Conducting Polymer Based Catalysts to Eliminate Pt for Dye Sensitized Solar Cell Applications. Eds.; Springer Nature: Cham, Switzerland, 2021, 349, 311-326, 978-3-030-62090-5_12.
- Wang, C.; Yuan, R.; Chai, Y.; Chen, S.; Hu, F.; Zhang, M. Simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan on gold nanoparticles/overoxidized-polyimidazole composite modified glassy carbon electrode. Anal. Chim. Acta 2012, 741, 15–20. Wang, C.; Yuan, R.; Chai, Y.; Chen, S.; Hu, F.; Zhang, M. Simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan on gold nanoparticles/overoxidized-polyimidazole composite modified glassy carbon electrode. Anal. Chim. Acta 2012, 741, 15–20, j.aca.2012.06.045.
- Luong, J.; Narayan, T.; Solanki, S.; Malhotra, B. Recent Advances of conducting polymers and their composites for electrochemical biosensing applications. J. Funct. Biomater. 2020, 11, 71. Luong, J.; Narayan, T.; Solanki, S.; Malhotra, B. Recent Advances of conducting polymers and their composites for electrochemical biosensing applications. J. Funct. Biomater. 2020, 11, 71, jfb11040071.
- Yang, L.; Liu, D.; Huang, J.; You, T. Simultaneous determination of dopamine, ascorbic acid and uric acid at electrochemically reduced graphene oxide modified electrode. Sens. Actuat. B-Chem. 2014, 193, 166–172. Yang, L.; Liu, D.; Huang, J.; You, T. Simultaneous determination of dopamine, ascorbic acid and uric acid at electrochemically reduced graphene oxide modified electrode. Sens. Actuat. B-Chem. 2014, 193, 166–172, j.snb.2013.11.104.
- Minta, D.; Moyseowicz, A.; Gryglewicz, S.; Gryglewicz, G. A promising Electrochemical Platform for dopamine and uric acid detection based on a a polyaniline/iron oxide-tin oxide/reduced graphene oxide ternary composite. Molecules 2020, 25, 5869. Minta, D.; Moyseowicz, A.; Gryglewicz, S.; Gryglewicz, G. A promising Electrochemical Platform for dopamine and uric acid detection based on a a polyaniline/iron oxide-tin oxide/reduced graphene oxide ternary composite. Molecules 2020, 25, 5869, molecules25245869.
- Feng, J.; Li, Q.; Cai, J.; Yang, T.; Chen, J.; Hou, X. Electrochemical detection mechanism of dopamine and uric acid on titanium nitride-reduced graphene oxide composite with and without ascorbic acid. Sens. Actuat. Chem-B 2019, 298, 126872. Feng, J.; Li, Q.; Cai, J.; Yang, T.; Chen, J.; Hou, X. Electrochemical detection mechanism of dopamine and uric acid on titanium nitride-reduced graphene oxide composite with and without ascorbic acid. Sens. Actuat. Chem-B 2019, 298, 126872, j.snb.2019.126872.
- Demirkan, B.; Bozkurt, S.; Cellat, K.; Arikan, K.; Yilmaz, M.; Savk, A.; Calimli, M.; Nas, M.; Atalar, M.; Alma, M.; et al. Palladium supported on polypyrrole/reduced graphene oxide nanoparticles for simultaneous biosensing application of ascorbic acid, dopamine, and uric acid. Sci. Rep. 2020, 10, 2946. Demirkan, B.; Bozkurt, S.; Cellat, K.; Arikan, K.; Yilmaz, M.; Savk, A.; Calimli, M.; Nas, M.; Atalar, M.; Alma, M.; et al.et al. Palladium supported on polypyrrole/reduced graphene oxide nanoparticles for simultaneous biosensing application of ascorbic acid, dopamine, and uric acid. Sci. Rep. 2020, 10, 2946, s41598-020-59935-y.
- Yola, M.L.; Atar, N. Functionalized graphene quantum dots with bi-metallic nanoparticles composite: Sensor application for simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan. J. Electrochem. Soc. 2016, 163, B718–B725. Yola, M.L.; Atar, N. Functionalized graphene quantum dots with bi-metallic nanoparticles composite: Sensor application for simultaneous determination of ascorbic acid, dopamine, uric acid and tryptophan. J. Electrochem. Soc. 2016, 163, B718–B725, 2.1191614jes.
- Loh, K.P.; Bao, Q.; Eda, G.; Chhowalla, M. Graphene oxide as a chemically tunable platform for optical applications. Nat. Chem. 2010, 2, 1015–1024. Loh, K.P.; Bao, Q.; Eda, G.; Chhowalla, M. Graphene oxide as a chemically tunable platform for optical applications. Nat. Chem. 2010, 2, 1015–1024, nchem.907.
- Li, Y.; Hu, Y.; Zhao, Y.; Shi, G.; Deng, L.; Hou, Y.; Qu, L. An electrochemical avenue to green luminescent graphene quantum dots as potential electron acceptors for photovoltaics. Adv. Mater. 2011, 23, 776–780. Li, Y.; Hu, Y.; Zhao, Y.; Shi, G.; Deng, L.; Hou, Y.; Qu, L. An electrochemical avenue to green luminescent graphene quantum dots as potential electron acceptors for photovoltaics. Adv. Mater. 2011, 23, 776–780, adma.201003819.
- Zhu, S.; Zhang, J.; Qiao, C.; Tang, S.; Li, Y.; Yuan, W.; Li, B.; Tian, L.; Liu, F.; Hu, R.; et al. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem. Commun. 2011, 47, 6858–6860. Zhu, S.; Zhang, J.; Qiao, C.; Tang, S.; Li, Y.; Yuan, W.; Li, B.; Tian, L.; Liu, F.; Hu, R.; et al.et al. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem. Commun. 2011, 47, 6858–6860, C1CC11122A.
- Gan, T.; Hu, C.; Sun, Z.; Hu, S. Facile synthesis of water-soluble fullerene-graphene oxide composites for electrodeposition of phosphotungstic acid-based electrocatalysts. Electrochim. Acta 2013, 111, 738–745. Gan, T.; Hu, C.; Sun, Z.; Hu, S. Facile synthesis of water-soluble fullerene-graphene oxide composites for electrodeposition ophosphotungstic acid-based electrocatalysts. Electrochim. Acta 2013, 111, 738–745, j.electacta.2013.08.059.
- Shan, C.; Yang, H.; Song, J.; Han, D.; Ivaska, A.; Niu, L. Direct Electrochemistry of glucose oxidase and biosensing for glucose based on graphene. Anal. Chem. 2009, 81, 2378–2382. Shan, C.; Yang, H.; Song, J.; Han, D.; Ivaska, A.; Niu, L. Direct Electrochemistry of glucose oxidase and biosensing for glucose based on graphene. Anal. Chem. 2009, 81, 2378–2382, ac802193c.
- Smaranda, I.; Benito, A.M.; Maser, W.K.; Baltog, I.; Baibarac, M. Electrochemical grafting of reduced graphene oxide with polydiphenylamine doped with heteropolyanions and its optical properties. J. Phys. Chem. C 2014, 118, 25704. Smaranda, I.; Benito, A.M.; Maser, W.K.; Baltog, I.; Baibarac, M. Electrochemical grafting of reduced graphene oxide with polydiphenylamine doped with heteropolyanions and its optical properties. J. Phys. Chem. C 2014, 118, 25704, jp507324r.
- Baibarac, M.; Ilie, M.; Baltog, I.; Lefrant, S.; Humbert, B. Infrared dichroism studies and anisotropic photoluminescence properties of poly(para-phenylene vinylene) functionalized reduced graphene oxide. RSC Adv. 2017, 7, 6931–6942. Baibarac, M.; Ilie, M.; Baltog, I.; Lefrant, S.; Humbert, B. Infrared dichroism studies and anisotropic photoluminescence properties of poly(para-phenylene vinylene) functionalized reduced graphene oxide. RSC Adv. 2017, 7, 6931–6942, C6RA26445J.
- Baibarac, M.; Daescu, M.; Fejer, S.N. Adsorption of 1, 4-phenylene diisocyanate onto the graphene oxide sheets functionalized with polydiphenylamine in doped state. Sci. Rep. 2019, 9, 11968. Baibarac, M.; Daescu, M.; Fejer, S.N. Adsorption of 1, 4-phenylene diisocyanate onto the graphene oxide sheets functionalized with polydiphenylamine in doped state. Sci. Rep. 2019, 9, 11968, s41598-019-48314-x.
- Baibarac, M.; Daescu, M.; Socol, M.; Bartha, C.; Negrila, C.; Fejer, S.N. Influence of reduced graphene oxide on the electropolymerization of 5-amino-1-naphthol and the interaction of 1, 4-phenylene diisothiocyanate with the poly(5-amino-1-naphtol)/reduced graphene oxide composite. Polymer 2020, 12, 1299. Baibarac, M.; Daescu, M.; Socol, M.; Bartha, C.; Negrila, C.; Fejer, S.N. Influence of reduced graphene oxide on the electropolymerization of 5-amino-1-naphthol and the interaction of 1, 4-phenylene diisothiocyanate with the poly(5-amino-1-naphtol)/reduced graphene oxide composite. Polymer 2020, 12, 1299, polym12061299.
- Lakshmi, D.; Whitcombe, M.; Davis, F.; Sharma, P.; Prasad, B.B. Electrochemical Detection of uric acid in mixed and clinical samples: A review. Electroanal 2011, 23, 305–320. Lakshmi, D.; Whitcombe, M.; Davis, F.; Sharma, P.; Prasad, B.B. Electrochemical Detection of uric acid in mixed and clinical samples: A review. Electroanal 2011, 23, 305–320, elan.201000525.
- Feng, X.-M.; Li, R.-M.; Ma, Y.-W.; Chen, R.-F.; Shi, N.-E.; Fan, Q.-L.; Huang, W. One-step electrochemical synthesis of graphene/polyaniline composite film and its applications. Adv. Funct. Mater. 2011, 21, 2989–2996. Feng, X.-M.; Li, R.-M.; Ma, Y.-W.; Chen, R.-F.; Shi, N.-E.; Fan, Q.-L.; Huang, W. One-step electrochemical synthesis ofgraphene/polyaniline composite film and its applications. Adv. Funct. Mater. 2011, 21, 2989–2996, adfm.201100038.
- Lim, C.; Chua, C.; Pumera, M. Detection of biomarkers graphene nanoplatelets and nanoribbons. Analyst 2014, 139, 1072–1080. Lim, C.; Chua, C.; Pumera, M. Detection of biomarkers graphene nanoplatelets and nanoribbons. Analyst 2014, 139, 1072–1080, C3AN01585H.
- Zhan, H.; Li, J.; Liu, Z.; Zheng, Y.; Jing, Y. A highly sensitive electrochemical OP biosensor based on electrodeposition of Au–Pd bimetallic nanoparticles onto a functionalized graphene modified glassy carbon electrode. Anal. Methods 2015, 7, 3903–3911. Zhan, H.; Li, J.; Liu, Z.; Zheng, Y.; Jing, Y. A highly sensitive electrochemical OP biosensor based on electrodeposition of Au–Pd bimetallic nanoparticles onto a functionalized graphene modified glassy carbon electrode. Anal. Methods 2015, 7, 3903–3911, C5AY00702J.
- Karimi-Maleh, H.; Arotiba, A.O. Simultaneous determination of cholesterol, ascorbic acid and uric acid as three essential biological compounds at a paste electrode modified with copper oxide decorated reduced graphene oxide nanocomposite and ionic liquid. J. Colloid. Interf. Sci. 2020, 560, 208–212. Karimi-Maleh, H.; Arotiba, A.O. Simultaneous determination of cholesterol, ascorbic acid and uric acid as three essential biological compounds at a paste electrode modified with copper oxide decorated reduced graphene oxide nanocomposite and ionic liquid. J. Colloid. Interf. Sci. 2020, 560, 208–212, j.jcis.2019.10.007.
- Kunpatee, K.; Traipop, S.; Chailapakul, O.; Chuanwatanakul, S. Simultaneous determination of ascorbic acid, dopamine and uric acid using graphene quantum dots/ionic liquid modified screen-printed carbon electrode. Sens. Actuat. B-Chem. 2020, 314, 128059. Kunpatee, K.; Traipop, S.; Chailapakul, O.; Chuanwatanakul, S. Simultaneous determination of ascorbic acid, dopamine and uric acid using graphene quantum dots/ionic liquid modified screen-printed carbon electrode. Sens. Actuat. B-Chem. 2020, 314, 128059, j.snb.2020.128059.
- Tian, J.; Deng, S.; Li, L.; Shan, D.; He, W.; Zhang, J.; Shi, Y. Bioinspired polydopamine as the scaffold for the active AuNPs anchoring and the simultaneously reduced graphene oxide: Characterization and the enhanced biosensing application. Biosens. Bioelectron. 2013, 49, 466–471. Tian, J.; Deng, S.; Li, L.; Shan, D.; He, W.; Zhang, J.; Shi, Y. Bioinspired polydopamine as the scaffold for the active AuNPs anchoring and the simultaneously reduced graphene oxide: Characterization and the enhanced biosensing application. Biosens. Bioelectron. 2013, 49, 466–471, j.bios.2013.06.009.
- Raj, V.; Madheswari, D.; MubarakAli, M. Chemical synthesis, characterization, and properties of conducting copolymers of imidazole and pyridine. J. Appl. Polym. Sci. 2012, 124, 1649–1658. Raj, V.; Madheswari, D.; MubarakAli, M. Chemical synthesis, characterization, and properties of conducting copolymers of imidazole and pyridine. J. Appl. Polym. Sci. 2012, 124, 1649–1658, app.35190.
- Yang, J.; Ramaraj, B.; Yoon, K. Preparation and characterization of superparamagnetic graphene oxide nanohybrids anchored with Fe3O4 nanoparticles. J. Alloys. Compd. 2014, 583, 128–133. Yang, J.; Ramaraj, B.; Yoon, K. Preparation and characterization of superparamagnetic graphene oxide nanohybrids anchored with Fe3O4 nanoparticles. J. Alloys. Compd. 2014, 583, 128–133, j.jallcom.2013.08.152.
- Xu, C.; Sun, J.; Gao, L. Synthesis of novel hierarchical graphene/polypyrrole nanosheet composites and their superior electrochemical performance. J. Mater. Chem. 2011, 21, 11253–11258. Xu, C.; Sun, J.; Gao, L. Synthesis of novel hierarchical graphene/polypyrrole nanosheet composites and their superior electrochemical performance. J. Mater. Chem. 2011, 21, 11253–11258, C1JM11275A.
- Kumar, G.; Kirubaharan, C.; Udhayakumar, S.; Ramachandran, K.; Karthikeyan, C.; Renganathan, R.; Nahm, K. Synthesis, structural, and morphological characterizations of reduced graphene oxide-supported polypyrrole anode catalysts for improved microbial fuel cell performances. ACS Sustain. Chem. Eng. 2014, 2, 2283–2290. Kumar, G.; Kirubaharan, C.; Udhayakumar, S.; Ramachandran, K.; Karthikeyan, C.; Renganathan, R.; Nahm, K. Synthesis, structural, and morphological characterizations of reduced graphene oxide-supported polypyrrole anode catalysts for improved microbial fuel cell performances. ACS Sustain. Chem. Eng. 2014, 2, 2283–2290, sc500244f.
- Harpale, K.; Bansode, S.; More, M. One-pot synthesis, characterization, and field emission investigations of composites of polypyrrole with graphene oxide, reduced graphene oxide, and graphene nanoribbons. J. Appl. Polym. Sci. 2017, 134, 1–8. Harpale, K.; Bansode, S.; More, M. One-pot synthesis, characterization, and field emission investigations of composites of polypyrrole with graphene oxide, reduced graphene oxide, and graphene nanoribbons. J. Appl. Polym. Sci. 2017, 134, 1–8, app.45170.
- Vellaichamy, B.; Periakaruppan, P.; Paulmony, T. Evaluation of a new biosensor based on in situ synthesized pPy-Ag-PVP nanohybrid for selective detection of dopamine. J. Phys. Chem. B 2017, 121, 1118–1127. Vellaichamy, B.; Periakaruppan, P.; Paulmony, T. Evaluation of a new biosensor based on in situ synthesized pPy-Ag-PVP nanohybrid for selective detection of dopamine. J. Phys. Chem. B 2017, 121, 1118–1127, acs.jpcb.6b11225.
- Hu, R.; Shao, D.; Wang, X. Graphene oxide/polypyrrole composites for highly selective enrichment of U (VI) from aqueous solutions. Polym. Chem. 2014, 5, 6207–6215. Hu, R.; Shao, D.; Wang, X. Graphene oxide/polypyrrole composites for highly selective enrichment of U (VI) from aqueous solutions. Polym. Chem. 2014, 5, 6207–6215, C4PY00743C.
- Bora, C.; Dolui, S. Fabrication of polypyrrole/graphene oxide nanocomposites by liqui/liqui interfacial polymerization and evaluation of their optical, electrical and electrochemical properties. Polymer 2012, 53, 923–932. Bora, C.; Dolui, S. Fabrication of polypyrrole/graphene oxide nanocomposites by liqui/liqui interfacial polymerization and evaluation of their optical, electrical and electrochemical properties. Polymer 2012, 53, 923–932, j.polymer.2011.12.054.
- Han, H.; Lee, H.; You, J.; Jeong, H.; Jeon, S. Electrochemical biosensor for simultaneous determination of dopamine and serotonin based on electrochemically reduced GO-porphyrin. Sens. Actuat. B-Chem. 2014, 190, 886–895. Han, H.; Lee, H.; You, J.; Jeong, H.; Jeon, S. Electrochemical biosensor for simultaneous determination of dopamine and serotonin based on electrochemically reduced GO-porphyrin. Sens. Actuat. B-Chem. 2014, 190, 886–895, j.snb.2013.09.022.
- Chen, L.; Guo, X.; Guo, B.; Cheng, S.; Wang, F. Electrochemical investigation of a metalloporphyrin-graphene composite modified electrode and its electrocatalysis on ascorbic acid. J. Electroanal. Chem. 2016, 760, 105–112. Chen, L.; Guo, X.; Guo, B.; Cheng, S.; Wang, F. Electrochemical investigation of a metalloporphyrin-graphene composite modified electrode and its electrocatalysis on ascorbic acid. J. Electroanal. Chem. 2016, 760, 105–112, j.jelechem.2015.11.018.
- Deng, M.; Yang, X.; Silke, M.; Qiu, W.; Xu, M.; Borghs, G.; Chen, H. Electrochemical deposition of polypyrrole/graphene oxide composite on microelectrodes towards tuning the electrochemical properties of neural probes. Sens. Actuat. B-Chem. 2011, 158, 176–184. Deng, M.; Yang, X.; Silke, M.; Qiu, W.; Xu, M.; Borghs, G.; Chen, H. Electrochemical deposition of polypyrrole/graphene oxide composite on microelectrodes towards tuning the electrochemical properties of neural probes. Sens. Actuat. B-Chem. 2011, 158, 176–184, j.snb.2011.05.062.
- Lu, G.; Zhou, M.; Lu, J.; Li, Z. Raman spectrum of mesosubstituted tetraphenylporphyrins. J. Jilin Univ. 2008, 2, 358–360. Available online: http://xuebao.jlu.edu.cn/lxb/EN/Y2008/V46/I02/358 (accessed on 1 November 2022).Lu, G.; Zhou, M.; Lu, J.; Li, Z. Raman spectrum of mesosubstituted tetraphenylporphyrins. J. Jilin Univ. 2008, 2, 358–360, 358.
- Li, D.; Liu, M.; Zhan, Y.; Su, Q.; Zhang, Y.; Zhang, D. Electrodeposited poly(3,4-ethylenedioxythiophene) doped with graphene oxide for the simultaneous voltametric determination of ascorbic acid, dopamine and uric acid. Microchim. Acta 2020, 187, 94. Li, D.; Liu, M.; Zhan, Y.; Su, Q.; Zhang, Y.; Zhang, D. Electrodeposited poly(3,4-ethylenedioxythiophene) doped with graphene oxide for the simultaneous voltametric determination of ascorbic acid, dopamine and uric acid. Microchim. Acta 2020, 187, 94, s00604-019-4083-4.
- Azman, N.; Lim, H.; Sulaiman, Y. Effect of electropolymerization potential on the preparation of PEDOT/graphene oxide hybrid material for supercapacitor application. Electrochim. Acta 2016, 188, 785–792. Azman, N.; Lim, H.; Sulaiman, Y. Effect of electropolymerization potential on the preparation of PEDOT/graphene oxide hybrid material for supercapacitor application. Electrochim. Acta 2016, 188, 785–792, j.electacta.2015.12.019.
- Lu, L.; Zhang, O.; Xu, J.; Wen, T.; Duan, X.; Yu, H.; Wu, L.; Nie, T. A facile one-step redox route for the synthesis of graphene/poly (3,4-ethylenedioxythiophene) nanocomposite and their applications in biosensing. Sens. Actuat. Chem-B 2013, 181, 567–574. Lu, L.; Zhang, O.; Xu, J.; Wen, T.; Duan, X.; Yu, H.; Wu, L.; Nie, T. A facile one-step redox route for the synthesis of graphene/poly (3,4-ethylenedioxythiophene) nanocomposite and their applications in biosensing. Sens. Actuat. Chem-B 2013, 181, 567–574, j.snb.2013.02.024.
- Yang, Y.; Li, S.; Yang, W.; Yuan, W.; Xu, J.; Jiang, Y. In situ polymerization deposition of porous conducting polymer on reduced graphene oxide for gas sensor. ACS Appl. Mater. Interfaces 2014, 6, 13807–13814. Yang, Y.; Li, S.; Yang, W.; Yuan, W.; Xu, J.; Jiang, Y. In situ polymerization deposition of porous conducting polymer on reduced graphene oxide for gas sensor. ACS Appl. Mater. Interfaces 2014, 6, 13807–13814, am5032456.
- Pandey, R.; Lakshminarayanan, V. Electro-oxidation of formic acid, methanol, and ethanol on electrodeposited Pd-polyaniline nanofiber films in acidic and alkaline medium. J. Phys. Chem. C 2009, 113, 21596–21603. Manivel, P.; Dhakshnamoorthy, M.; Balamurugan, A.; Ponpandian, N.; Mangalaraja, D.; Viswanathan, C. Conducting polyaniline-graphene oxide fibrous nanocomposites: Preparation, characterization and simultaneous electrochemical detection of ascorbic acid, dopamine and uric acid. RSC Adv. 2013, 3, 14428–14437, C3RA42322K.
- Chen, X.; Li, D.; Ma, W.; Yang, T.; Zhang, Y.; Zhang, D. Preparation of a glassy carbon electrode modified with reduced graphene oxide and overoxidized electropolymerized polypyrrole, and its application to the determination of dopamine in the presence of ascorbic acid and uric acid. Microchim. Acta 2019, 186, 407. Chen, G.; Shau, S.; Juang, T.; Lee, R.; Chen, C.; Suen, S.; Jeng, R. Single-layered graphene oxide nanosheet/polyaniline hybrids fabricated through direct molecular exfoliation. Langmuir 2011, 27, 14563–14569, la203253m.
- Tukimin, N.; Abdullah, J.; Sulaiman, Y. Electrodeposition of poly(3,4-ethylenedioxytiophene)/reduced graphene oxide/manganese dioxide for simultaneous detection of uric acid, dopamine and ascorbic acid. J. Electroanal. Chem. 2018, 820, 74–81. Wang, H.; Hao, Q.; Yang, X.; Lu, L.; Wang, X. Effect of graphene oxide on the properties of its composite with polyaniline. ACS Appl. Mater. Interfaces 2010, 2, 821–828, am900815k.
- Tiwari, I.; Gupta, M.; Pandey, C.; Mishra, V. Gold nanoparticle decorated graphene sheet-polypyrrole based nanocomposite: Its synthesis, characterization and genosensing application. Dalton Trans. 2015, 44, 15557–15566. Zhang, F.; Gu, S.; Ding, Y.; Zhou, L.; Zhang, Z.; Li, L. Electrooxidation and determination of cefotaxime on Au nanoparticles/poly(L-arginine) modified carbon paste electrode. J. Electroanal. Chem. 2013, 698, 25–30, j.jelechem.2013.03.010.
- Manivel, P.; Dhakshnamoorthy, M.; Balamurugan, A.; Ponpandian, N.; Mangalaraja, D.; Viswanathan, C. Conducting polyaniline-graphene oxide fibrous nanocomposites: Preparation, characterization and simultaneous electrochemical detection of ascorbic acid, dopamine and uric acid. RSC Adv. 2013, 3, 14428–14437. Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited II . J. Phys. Chem. B 1998, 102, 4477–4482, jp9731821.
- Gao, F.; Cai, X.; Wang, X.; Gao, C.; Liu, S.; Gao, F.; Wang, Q. Highly sensitive and selective detection of dopamine in the presence of ascorbic acid at graphene oxide modified electrode. Sens. Actuat B-Chem. 2013, 186, 380–387. Prasert, K.; Sutthibutpong, T. Unveiling the fundamental mechanisms of graphene oxide selectivity on the ascorbic acid, dopamine, and uric acid by Density Functional Theory Calculations and Charge Population Analysis. Sensors 2021, 21, 2773, s21082773.
- Li, F.; Chai, J.; Yang, H.; Han, D.; Niu, L. Synthesis of Pt/ionic liquid/graphene nanocomposite and its simultaneous determination of ascorbic acid and dopamine. Talanta 2010, 81, 1063–1068. Domancich, N.; Fernandez, A.; Meier, L.; Fuente, S.; Castellani, N. DFT study of graphene oxide reduction by a dopamine species. Mol. Phys. 2020, 118, e1637029, 00268976.2019.1637029.
- Rahman, M.; Lopa, N.; Kim, K.; Lee, J.-J. Selective detection of L-tyrosine in the presence of ascorbic acid, dopamine, and uric acid at poly (thionine)-modified glassy carbon electrode. J. Electroanal. Chem. 2015, 754, 87–93. Pandey, R.; Lakshminarayanan, V. Electro-oxidation of formic acid, methanol, and ethanol on electrodeposited Pd-polyaniline nanofiber films in acidic and alkaline medium. J. Phys. Chem. C 2009, 113, 21596–21603, jp908239m.
- Li, X.-B.; Rahman, M.; Xu, G.-R.; Lee, J.-J. Highly sensitive and selective detection of dopamine at poly (chromotrope 2B)-modified glassy carbon electrode in the presence of uric acid and ascorbic acid. Electrochim. Acta 2015, 173, 440–447. Chen, X.; Li, D.; Ma, W.; Yang, T.; Zhang, Y.; Zhang, D. . Preparation of a glassy carbon electrode modified with reduced graphene oxide and overoxidized electropolymerized polypyrrole, and its application to the determination of dopamine in the presence of ascorbic acid and uric acid. Microchim. Acta 2019, 186, 407, s00604-019-3518-2.
- Guo, W.; Xu, L.; Li, F.; Xu, B.; Yang, Y.; Liu, S.; Sun, Z. Chitosan-assisted fabrication and electrocatalytic activity of the composite film electrode of heteropolytungstate/carbon nanotubes. Electrochim. Acta 2010, 55, 1523–1527. Tukimin, N.; Abdullah, J.; Sulaiman, Y. Electrodeposition of poly(3,4-ethylenedioxytiophene)/reduced graphene oxide/manganese dioxide for simultaneous detection of uric acid, dopamine and ascorbic acid. J. Electroanal. Chem. 2018, 820, 74–81, j.jelechem.2018.04.065.
- Juan, L.; Xiaoli, Z. Fabrication of poly(aspartic acid)-Nanogold modified electrode and its application for simultaneous determination of dopamine, ascorbic acid, and uric acid. Am. J. Anal. Chem. 2012, 3, 195–203. Tiwari, I.; Gupta, M.; Pandey, C.; Mishra, V. Gold nanoparticle decorated graphene sheet-polypyrrole based nanocomposite: Its synthesis, characterization and genosensing application. Dalton Trans. 2015, 44, 15557–15566, C5DT01193K.
- Ramirez-Silva, M.; Corona-Avendano, S.; Alarcon-Angeles, G.; Palomar-Parave, M.; Romero-Romo, M.; Rojas-Hernandez, A. Construction of Supramolecular Systems for the selective and quantitative determination of dopamine in the presence of ascorbic acid. Procedia Chem. 2014, 12, 55–61. Gao, F.; Cai, X.; Wang, X.; Gao, C.; Liu, S.; Gao, F.; Wang, Q. Highly sensitive and selective detection of dopamine in the presence of ascorbic acid at graphene oxide modified electrode. Sens. Actuat B-Chem. 2013, 186, 380–387, j.snb.2013.06.020.
- Zhang, F.; Gu, S.; Ding, Y.; Zhang, Z.; Li, L. A novel sensor based on electropolymerization of beta-cyclodextrin and L-arginine on carbon paste electrode for determination of fluoroquinolones. Anal. Chim. Acta 2013, 770, 53–61. Li, F.; Chai, J.; Yang, H.; Han, D.; Niu, L. Synthesis of Pt/ionic liquid/graphene nanocomposite and its simultaneous determination of ascorbic acid and dopamine. Talanta 2010, 81, 1063–1068, j.talanta.2010.01.061.
- Rahman, M.; Lopa, N.; Kim, K.; Lee, J.-J. Selective detection of L-tyrosine in the presence of ascorbic acid, dopamine, and uric acid at poly (thionine)-modified glassy carbon electrode. J. Electroanal. Chem. 2015, 754, 87–93, j.jelechem.2015.06.018.
- Li, X.-B.; Rahman, M.; Xu, G.-R.; Lee, J.-J. Highly sensitive and selective detection of dopamine at poly (chromotrope 2B)-modified glassy carbon electrode in the presence of uric acid and ascorbic acid. Electrochim. Acta 2015, 173, 440–447, j.electacta.2015.05.062.
- Guo, W.; Xu, L.; Li, F.; Xu, B.; Yang, Y.; Liu, S.; Sun, Z. Chitosan-assisted fabrication and electrocatalytic activity of the composite film electrode of heteropolytungstate/carbon nanotubes. Electrochim. Acta 2010, 55, 1523–1527, j.electacta.2009.10.003.
- Juan, L.; Xiaoli, Z. Fabrication of poly(aspartic acid)-Nanogold modified electrode and its application for simultaneous determination of dopamine, ascorbic acid, and uric acid. Am. J. Anal. Chem. 2012, 3, 195–203, ajac.2012.33028.
- Ramirez-Silva, M.; Corona-Avendano, S.; Alarcon-Angeles, G.; Palomar-Parave, M.; Romero-Romo, M.; Rojas-Hernandez, A. Construction of Supramolecular Systems for the selective and quantitative determination of dopamine in the presence of ascorbic acid. Procedia Chem. 2014, 12, 55–61, j.proche.2014.12.041.
- Zhang, F.; Gu, S.; Ding, Y.; Zhang, Z.; Li, L. A novel sensor based on electropolymerization of beta-cyclodextrin and L-arginine on carbon paste electrode for determination of fluoroquinolones. Anal. Chim. Acta 2013, 770, 53–61, j.aca.2013.01.052.
More