Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Dean Liu and Version 2 by Dean Liu.
EC (Endothelial Cell) metabolism is confined to glucose, FAs, and amino acids (AAs), the three major substrates for adenosine triphosphate (ATP) production and biomass production in ECs, which have been widely studied and summarized.
- endothelium
- vascular biology
- lipid
- glycolysis
1. Endothelial Cell Glucose and Amino Acid Metabolism
Glycolysis is the main energy resource in cultured ECs, with higher rates of glycolysis and glucose consumption [1]. ECs prefer to utilize glycolysis instead of oxidative metabolism because the ECs need to maintain reactive oxygen species (ROS) levels in control [2] and enhance the diffusion of oxygen to perivascular cells [2][3]. Additional reasons are that glycolysis produces faster kinetic ATP under pro-angiogenic signals during angiogenesis [1], which is essential for ECs’ rapid proliferation and migration [1][4]. In addition to glucose-derived carbons, ECs also utilize glutamine to sustain proliferation and vascular expansion [5][6]. Glutamine, the most abundant circulating nonessential amino acid (NEAA), can supply 30% of the tricarboxylic acid (TCA) carbons, comparable to glycolysis and FAO-derived carbon [7]. Depletion of either glutamine or arginine makes ECs vulnerable to ROS-induced damage during proliferation and migration [6].
2. Endothelial Cell Fatty Acids (FAs) Metabolism
FA metabolism involves multiple processes including FA uptake and storage, FA transport, FA oxidation, and FA synthesis. FA metabolism is vitally important to sustain the function of organs such as the heart, skeletal muscle, and adipose tissue [8]. Heart and skeletal muscles utilize FAs as their top source for ATP production and therefore require an efficient supply system of FAs [9]. Uptake and transport of FAs by ECs are extremely important to numerous cellular processes, including membrane synthesis, intracellular signal transduction, ATP generation, protein posttranslational modifications, and metabolic gene transcriptional regulation in these high energy-demand organs [10]. This process requires the transport of FA across the EC barrier into the perivascular cells [11] (Figure 1). In addition, ECs can metabolize and synthesize FAs for maintaining vascular homeostasis.
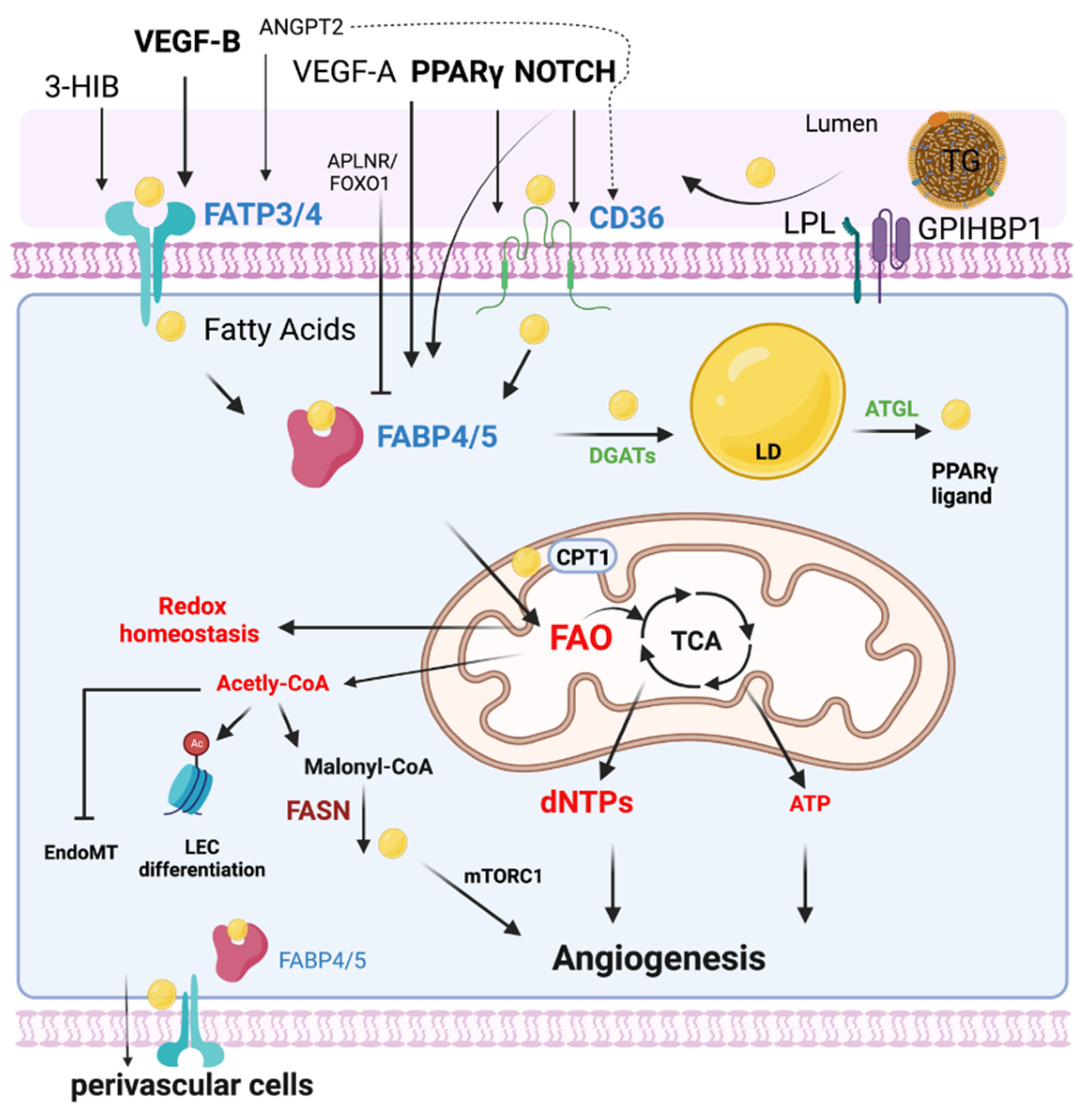
Figure 1. EC FA metabolism. Fatty acids (FAs) are transported in the form of triglyceride (TG)-rich lipoproteins, which are released by lipoprotein lipase (LPL) and GPIHBP1 in the luminal side of ECs. Free FAs are transported across EC membranes by fatty acid transporter protein 3 and 4 (FATP3/4) and CD36. Intracellular FAs transportation is mediated by FABP3 and FABP4. Intracellular FAs could be synthesized by FA synthase (FASN) from Malonyl-CoA and affect mTORC1 activity for angiogenesis. DAGTs and ATGL are key enzymes regulating lipid droplet (LD) storage and lipolysis. FAs are transported into mitochondria via CPT1 for FA oxidation (FAO). Quiescent ECs utilize FAO to maintain redox homeostasis. FAO is required for the generation of dNTPs for EC spouting and angiogenesis. In certain conditions such as glucose depletion, FAO produces ATP in ECs. FAO also produces acetyl-CoA for epigenetic regulation, inhibiting EndoMT. The efflux of FAs to the perivascular cells could be mediated by FABP4/5 and FATP3/4. Many key endothelial signaling including VEGF, NOTCH, and PPAR γ control EC FA transport via regulating FATPs, FABPs, and CD36.
3. Fatty Acid Metabolism in Pulmonary Hypertension
Given multiple molecules and their complex regulation network involved in FA metabolism in ECs, any abnormality in these regulations will lead to a pathological state and even develop into a disease. Here, researchers will only discuss the role of FA metabolism and its related pathway in pulmonary hypertension (PH) (Figure 2).
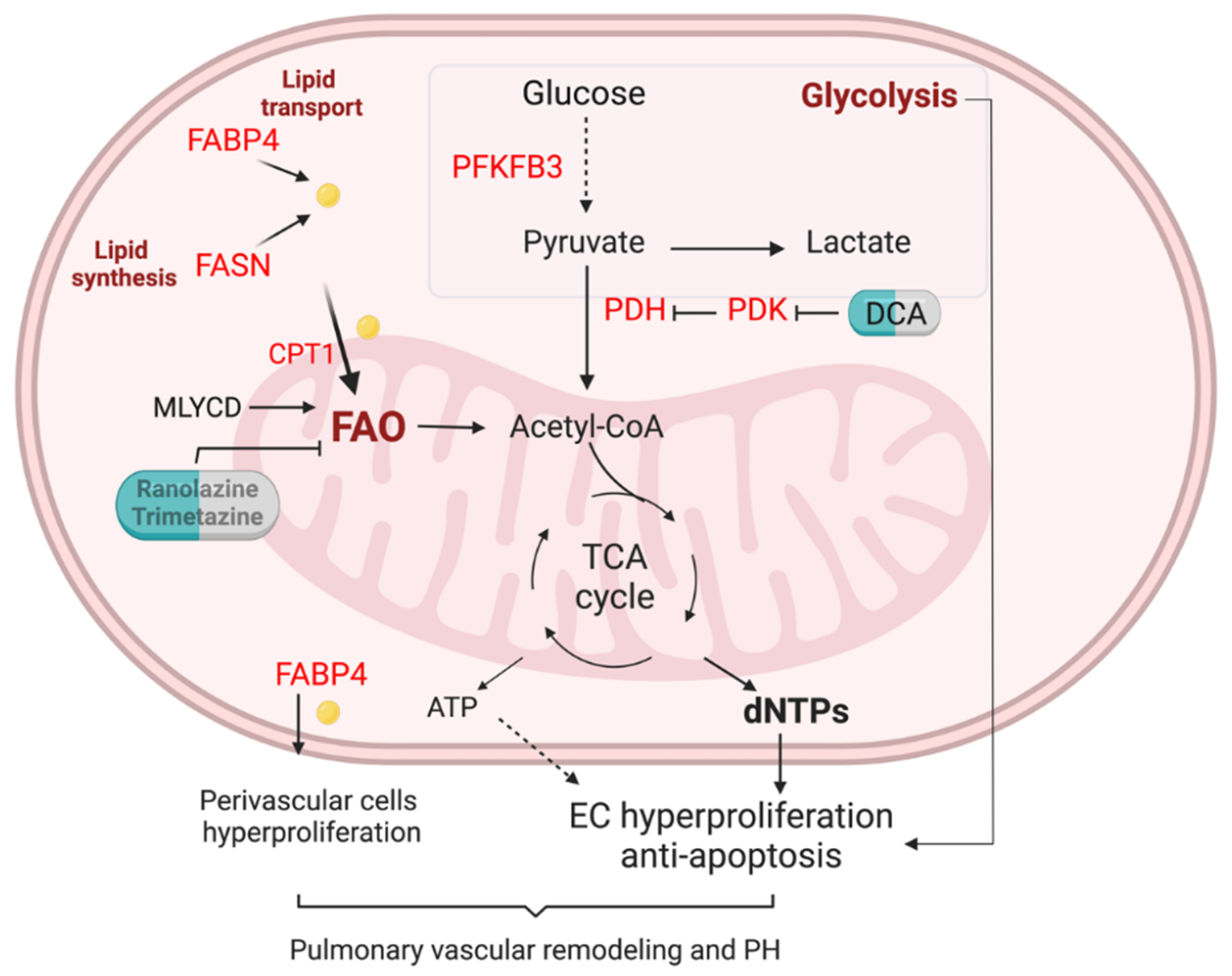
Figure 2. FA metabolism in the lung EC of pulmonary hypertension. Enhanced glycolysis is evident in PAH ECs. Lipid transport via FABP4, lipid synthesis via fatty acid synthase (FASN), and fatty acid oxidation (FAO) are unregulated in pulmonary arterials of PAH patients. Elevation of FA could increase mitochondria FAO, contributing to the generation of dNTPs and ATP. Enhanced FAO and glycolysis lead to PAH EC hyperproliferation and anti-apoptotic phenotypes. Upregulation of FAPB4 could also increase FA efflux to perivascular cells such as pulmonary arterial smooth muscle cells and fibroblasts, and promote pulmonary vascular remodeling and PH.
Pulmonary arterial hypertension (PAH), or group 1 PH, is an incurable lung disease, characterized by increased resistance in the pulmonary vascular system, which will finally lead to elevated resting pulmonary artery pressure and end by right ventricular failure [12]. Accumulating evidence has demonstrated that EC dysfunction plays a crucial role in the initiation and progression of PAH, manifested by increased susceptibility to injury and enhanced proliferation, contributing to the formation of plexiform lesions [13][14][15][16]. Indeed, most PAH mutation-causing genes are mainly expressed in the lung ECs based on the human lung single-cell RNA sequencing analysis [17].
To date, increasing evidence indicates that EC metabolism dysfunction is closely associated with the pathogenesis of PAH [18][19][20]. The major observed metabolic alteration of EC metabolic change in PAH has increased glycolysis [18]. Glucose metabolic activities were higher in the lungs of PAH patients than in healthy individuals determined by in vivo fluorodeoxyglucose (FDG)-positron emission tomography (PET) scans [21][22]. Healthy ECs generate 85% of their energy from glycolysis. The glycolytic rate of PAH ECs is even greater compared to healthy ECs [21][22], suggesting that PAH ECs exhibit a further shift to aerobic glycolysis. Pulmonary vascular ECs sustain the proliferative and anti-apoptotic phenotypes depending on enhanced glycolysis. Mice with EC-specific deletion of PFKFB3, a key regulator of glycolysis, slowed PH development [23].
FA metabolism abnormality has been observed in PH. Metabolites profiling of the plasma from patients with PAH and healthy controls showed the dysregulated metabolic pathways. Metabolites representing lipid metabolism and fatty acid were closely associated with PAH [24]. Circulating levels of FABP4 were significantly elevated in PAH patients identified by plasma proteome profiling [24]. Lipid deposition is increased in the lungs of PH patients compared to control lungs [25]. FASN was upregulated in hypoxic pulmonary arterial smooth muscle cells (PASMCs) and pulmonary arterial ECs (PAECs), and monocrotaline (MCT)-treated rats (a widely used PH model) [26][27]. FASN inhibition using the C75 compound decreased right ventricular pressure, right heart hypertrophy, pulmonary vascular remodeling, and endothelial dysfunction in MCT-exposed rats [27] and hypoxic mice [28].
Previous studies showed that non-esterified free fatty acids and acylcarnitines in the circulation are upregulated in patients with PAH compared to healthy controls [29][30]. Using a combination of high-throughput liquid-and-gas-chromatography-based mass spectrometry analysis on human PAH lung, Zhao et al. observed that there were increased long- and medium-chain free fatty acid products accumulated in PAH tissues compared to control lung, a reflection of mitochondria β-oxidation [31]. They also demonstrated an increase of omega-oxidation in fatty acids and upregulation of lipid oxidation in the lung of PAH [31]. Recent studies also demonstrated the upregulation of FA uptake, processing, and β-oxidation-related genes in the laser-dissected pulmonary arteries from idiopathic PAH (IPAH) patients [32]. The unpublished data showed that PAECs isolated from IPAH patients exhibited increased exogenous FAO compared to PAECs from failed donors using Seahorse XF Palmitate Oxidation Stress assay, suggesting upregulation of FAO in PAH EC. Other evidence showed that inhibition of FAO via genetic deletion of malonyl-CoA decarboxylase (MYLCD) in the whole body protected animal models from developing PH in mice [33]. Genetic knockdown of Cpt1a in mice or pharmacological inhibition (Oxfenicine) of Cpt1b in rats attenuated pulmonary vascular remodeling and PH [32].
In contrast, other studies showed a decrease in carnitine acyltransferase (CrAT), indicative of the reduction in FAO, in the endothelium of congenital heart disease and the PH lamb model [34]. Metabolomic analysis of bone morphogenetic protein receptor type 2 (BMPR2) mutations, which account for 80% of heritable PAH, in human pulmonary EC reveals the downregulation of the carnitine and FAO pathway [35].
PAH patients mostly die because of right heart failure. However, the role of FAO in RV failure is also controversial. Recent studies have demonstrated that intracellular lipid accumulation and the reduction of FAO are features of right heart failure secondary to PAH [29][30]. Human right ventricular (RV) long-chain FAs, myocardial triglyceride content, and ceramide were increased and long-chain acylcarnitines were markedly reduced in PAH versus controls [29]. Activation of FAO using PPAR-γ agonist pioglitazone or metformin or L-carnitine decreased lipid accumulation and improved right heart function in PH animal models [30][36][37]. However, another group demonstrated that partial inhibition of FAO using Trimetazidine restored pyruvate dehydrogenase (PDH) activity and glucose oxidation, and improved RV function in pulmonary artery banding rats [38] via manipulating Randle’s cycle, for example, inhibiting FAO increases glucose oxidation, or vice versa [38].
Taken together, the role of endothelial FA metabolism in the pathogenesis of PAH and RV failure is complicated and needed for further investigation.
References
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663.
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647.
- Helmlinger, G.; Endo, M.; Ferrara, N.; Hlatky, L.; Jain, R.K. Formation of endothelial cell networks. Nature 2000, 405, 139–141.
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228.
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333.
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352.
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197.
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503.
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921.
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857.
- Hülsmann, W.C.; Dubelaar, M.L. Aspects of fatty acid metabolism in vascular endothelial cells. Biochimie 1988, 70, 681–686.
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492.
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458.
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802.
- Liu, B.; Peng, Y.; Yi, D.; Machireddy, N.; Dong, D.; Ramirez, K.; Dai, J.; Vanderpool, R.; Zhu, M.M.; Dai, Z.; et al. Endothelial PHD2 deficiency induces nitrative stress via suppression of caveolin-1 in pulmonary hypertension. Eur. Respir. J. 2022, 60, 2102643.
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957.
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625.
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094.
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in Pulmonary Hypertension. Annu. Rev. Physiol. 2021, 83, 551–576.
- Shi, X.F.; Su, Y.C. Vascular Metabolic Mechanisms of Pulmonary Hypertension. Curr. Med. Sci. 2020, 40, 444–454.
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347.
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372.
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403.
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z.; et al. Metabolomics reveals metabolite changes of patients with pulmonary arterial hypertension in China. J. Cell Mol. Med. 2020, 24, 2484–2496.
- Umar, S.; Ruffenach, G.; Moazeni, S.; Vaillancourt, M.; Hong, J.; Cunningham, C.; Cao, N.; Navab, S.; Sarji, S.; Li, M.; et al. Involvement of Low-Density Lipoprotein Receptor in the Pathogenesis of Pulmonary Hypertension. J. Am. Heart Assoc. 2020, 9, e012063.
- Singh, N.; Singh, H.; Jagavelu, K.; Wahajuddin, M.; Hanif, K. Fatty acid synthase modulates proliferation, metabolic functions and angiogenesis in hypoxic pulmonary artery endothelial cells. Eur. J. Pharmacol. 2017, 815, 462–469.
- Singh, N.; Manhas, A.; Kaur, G.; Jagavelu, K.; Hanif, K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br. J. Pharmacol. 2016, 173, 2030–2045.
- Hou, C.; Chen, J.; Zhao, Y.; Niu, Y.; Lin, S.; Chen, S.; Zong, Y.; Sun, X.; Xie, L.; Xiao, T. The Emerging Role of Fatty Acid Synthase in Hypoxia-Induced Pulmonary Hypertensive Mouse Energy Metabolism. Oxidative Med. Cell. Longev. 2021, 2021, 9990794.
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944.
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334.
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727.
- Lee, M.H.; Sanders, L.; Kumar, R.; Hernandez-Saavedra, D.; Yun, X.; Ford, J.A.; Perez, M.J.; Mickael, C.; Gandjeva, A.; Koyanagi, D.E.; et al. Contribution of fatty acid oxidation to the pathogenesis of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L355–l371.
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58.
- Sun, X.; Sharma, S.; Fratz, S.; Kumar, S.; Rafikov, R.; Aggarwal, S.; Rafikova, O.; Lu, Q.; Burns, T.; Dasarathy, S.; et al. Disruption of endothelial cell mitochondrial bioenergetics in lambs with increased pulmonary blood flow. Antioxid. Redox Signal. 2013, 18, 1739–1752.
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ. 2012, 2, 201–213.
- Agrawal, V.; Hemnes, A.R.; Shelburne, N.J.; Fortune, N.; Fuentes, J.L.; Colvin, D.; Calcutt, M.W.; Talati, M.; Poovey, E.; West, J.D.; et al. l-Carnitine therapy improves right heart dysfunction through Cpt1-dependent fatty acid oxidation. Pulm. Circ. 2022, 12, e12107.
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303.
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43.
More