Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Qingguo Ma and Version 2 by Dean Liu.
Cycloketones can be oxidized to lactones using molecular oxygen, peroxy acids, or hydrogen peroxide as an oxidant. Hydrogen peroxide is one of the environmental oxidants. Because of the weak oxidation ability of hydrogen peroxide, Bronsted acids and Lewis acids are used as catalysts to activate hydrogen peroxide or the carbonyl of ketones to increase the nucleophilic performance of hydrogen peroxide. The catalytic mechanisms of Bronsted acids and Lewis acids differ in the Baeyer–Villiger oxidation of cyclohexanone with an aqueous solution of hydrogen peroxide as an oxidant.
- Lewis acid
- cycloketone
- lactone
1. Homogeneous Systems
Markiton M. studied a range of metal triflates in the BV oxidation of 2-adamantanone. The results showed activity of Sn(OTf)2 was higher than other metal triflates. Both the conversion of 2-adamantanone and the yield of lactone were higher than 99% (0.67 mmol 2-adamantanone, 1.34 mmol 30 wt% H2O2, 10 mol% Sn(OTf)2, 6 mL toluene, 70 °C, 30 min). To increase the catalytic activity of Sn(OTf)2, it was immobilized on multi-walled carbon nanotubes (MWCNTs), and then hydrolyzed to triflic acid in the BV oxidation. The catalytic activity of the recycled catalyst decreased. The participation of both Lewis and Bronsted acid sites in the conversion of the ketone was postulated [1][26].
Ionic liquids were easily leached from porous solids during the reaction. The supported ionic liquid catalyst was prepared by a ship-in-a-bottle technique or organic-additive-instant seed technology. Modi [2][27] synthesized [BMIM]BF4@Co/ZSM-5 and [BMIM]BF4@Co/HY. They were tested in the BV oxidation of cyclohexanone using 30% H2O2 as an oxidant under solvent-free conditions. [BMIM]BF4@Co/HY showed high catalytic activity with the conversion of cyclohexanone being 54.88%, and the ε-caprolactone selectivity 86.36% (50 mg [BMIM]BF4@Co/HY, 0.03 mol cyclohexanone, 0.06 mol 30 wt% H2O2, 75 °C, 6 h). [BMIM]BF4@Co/HY interacts with carbonyl and hydrogen peroxide simultaneously. The nucleophile BF4− plays an essential role in the catalytic reaction. The supported ionic liquid catalysts prepared by the ship-in-a-bottle technique or organic additive-instant seed methods were unstable [3][4][28,29]. Although the selectivity of ε-caprolactone was maintained at about 80%, the activity loss is considerable after six recycling cycles.
A series of oxo-rhenium complexes were prepared by reacting with Re2O7 using PTA, mPTA, HMT, Tpm, and Li(Tpms) as raw materials, respectively (Figure 1) [5][30]. These oxo-rhenium complexes’ catalytic activity was studied toward the BV oxidation of cyclic and linear ketones. These prepared oxo-rhenium complexes exhibit higher catalytic activity in the oxidation of ketones with H2O2 than the simple rhenium oxides K[ReO4] and Re2O7. The catalytic performances of oxo-rhenium complexes are shown Table 1. Although the oxo-rhenium complexes have good water solubility and are stable in an aqueous solution, the selectivity of the corresponding product ester is poor (<36%) when catalyzing the BV reaction of ketones.
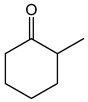
Table 1. Catalytic performance of Re complexes for the oxidation of cyclic ketones to lactones.
| Ketone | Product | Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|
 |
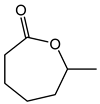 |
a | 66 | 45 |
| b | 85 | 36 | ||
| c | 68 | 22 | ||
| d | 75 | 21 | ||
| e | 67 | 16 | ||
| f | 70 | 10 | ||
| Re2O7 | 60 | 11 | ||
 |
 |
a | 59 | 20 |
| b | 37 | 43 | ||
| d | 38 | 26 | ||
| Re2O7 | 55 | 5 | ||
 |
 |
a | 63 | 37 |
| b | 58 | 41 | ||
| d | 67 | 51 |
1.7 μmol catalyst (The structures of catalysts a, b, c, d, e, and f are shown in Figure 1), n(ketone)/n(H2O2) = 1.7 mmol/1.7 mmol, 3 mL 1,2-dichloroethane, 70 °C, 6 h.
Frisone studied the BV oxidation of cyclic ketones with H2O2 catalyzed by platinum (II) complexes (Figure 2) [6][31]. (P-P)Pt(CF3)X (P-P = diphosphine; X = solvent, −OH) showed high activity for the BV oxidation of ketones using a commercial 32 wt% H2O2 solution. The conversion of cyclic ketones to the corresponding lactones is shown in Table 2. The solubility of the bis-cationic PtII catalyst in water led to hydrolysis with the release of one equivalent of H+. For more acid-sensitive lactones, the formation of the corresponding organic acid was increased because of H+-catalyzed hydrolysis of the intermediate lactone in water, higher temperatures, and the high catalyst loading result in lower yields of lactones.
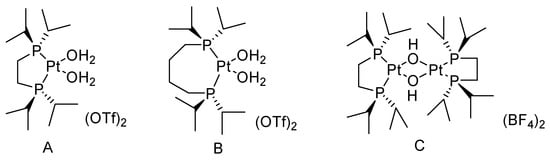
Table 2. Catalytic performance of (P-P)Pt(CF3)X for the oxidation of cyclic ketones to lactones.
| Ketone | Product | Yield (mmol) | Time | Solvent |
|---|---|---|---|---|
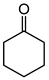 |
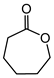 |
1.94 | 35 min | - |
| 1.47 | 25 min | CH2Cl2 | ||
| 2.88 | 2.8 h | CH2Cl2 | ||
| 1.98 | 10 min | - | ||
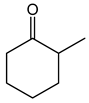 |
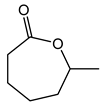 |
2.7 | 49 min | - |
| 4.84 | 4 h | CH2Cl2 | ||
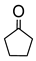 |
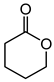 |
3.05 | 1.8 h | - |
| 1.62 | 5 h | THF | ||
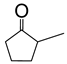 |
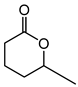 |
0.77 | 51 min | - |
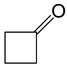 |
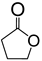 |
15.0 | 8 min | - |
| 37.1 | 2.75 h | - |
Reaction conditions: 0.045 mmol Pt; 45 mmol ketone; 22.5 mmol H2O2; N2 l atm; 1 mL solvent (where present).
2. Heterogeneous Systems
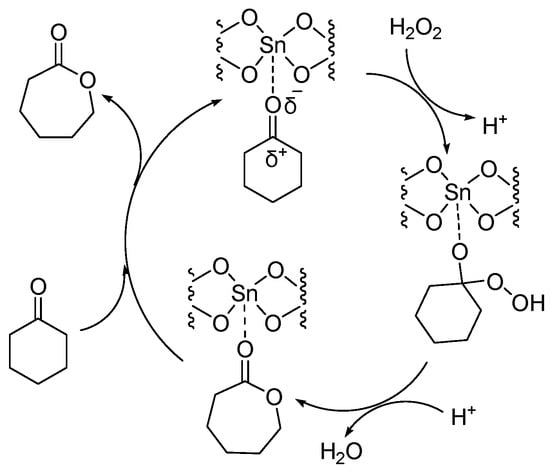
Sn-containing materials were extensively used as a highly active Lewis acid heterogeneous catalyst [7][8][9][10][32,33,34,35]. Because of the large specific surface area and various channel structures, mesoporous materials have attracted much attention in the field of catalysis. Corma A. synthesized a catalyst (Sn-zeolite beta) that had discrete and isolated Lewis sites [11][36]. Sn was incorporated into the zeolite framework as tetrahedrally coordinated Sn. The chemoselectivity of Sn-zeolite beta in the BV oxidation of ketones was high when using H2O2 as an oxidant. The selectivity for lactones was 100%. The mechanism of the BV reaction of cyclohexanone catalyzed by Sn-zeolite beta using aqueous hydrogen peroxide as an oxidant is shown in Figure 3. The oxygen atom in the ketone is coordinated with the Sn (Lewis acid center). Then it was easy for the oxygen atom of hydrogen peroxide to attack the carbon atom of carbonyl. The catalytic activity of Sn-zeolite beta in the BV oxidation of adamantanone and cyclohexanone was higher than the best homogeneous catalytic system. In addition, Sn-zeolite beta was filtered out and reused four times without special treatment, and its activity did not decrease in the BV oxidation of adamantanone and cyclohexanone. However, the preparation of Sn-zeolite beta was complex. The crystallization of Sn-zeolite beta takes 20 days at 140 °C in a stainless steel autoclave.
Different tin contents of Sn-Beta zeolites were synthesized by aerosol-seed-assisted hydrothermal methods. This method reduced the amount of template and mineralizer and the reaction time. The optimal condition was obtained through several synthesis parameters (1SiO2: 0.4TEAOH: 0.4HF: 5.6H2O with 3 wt% of seeds, 423 k, 6–9 days). The synthesized catalyst of Sn-Beta zeolite was tested in the BV oxidation of cyclohexanone using H2O2 as an oxidant. With the increase in tin content, the conversion of ketone increased, and the selectivity of ε-caprolactone decreased. The amount of the extra framework Sn sites increased with the increase in tin content, which may cause the oxidation of ε-caprolactone to acid [12][37].
Different Sn-zeolites (Sn-MCM-68, Sn-BEA, Sn-MWW, Sn-MFI) were synthesized and evaluated in the BV oxidation of 2-adamantanone. By comparison, Sn-MCM-68 possesses more catalytic activity because of its three-dimensional channels and relatively higher hydrophobicity [13][38]. After the reaction, Sn-MCM-68 and Sn-BEA were separated, washed with chlorobenzene, and calcined. The catalytic activity of the regenerated catalyst was comparable to the fresh catalyst.
Han Y. synthesized a series of Mg-Sn-W composite oxides with different W contents. These catalysts were tested in the BV oxidation of cyclohexanone. The tests of BV oxidation with Mg-Sn-W oxide catalysts showed high activity, and the selectivity of caprolactone approached 90%. The tungsten oxide was the main active component. Mg-Sn-W oxide was separated by filtration and used again immediately. The catalytic activity decreased significantly (conversion of cyclohexanone is 37%, selectivity of ε-caprolactone is 75%). Mg-Sn-W oxide was separated, washed with water and ethanol, and calcined at 400 °C. Although the structure of the recycled catalyst was not destroyed, the catalytic activity was still slightly worse than the fresh catalysts. (conversion: 59% (second), 55% (third), 50% (fourth); selectivity: 83% (second), 80% (third), 76% (fourth)) [14][39]. A series of magnesium–aluminum hydrotalcite-like compounds were prepared to catalyze the BV oxidation of cyclohexanone using H2O2 as oxidation. The results showed that the crystal size affected the catalytic activity. Enhanced hydrophilicity of poorly crystalline HT samples facilitates the approach and activation of H2O2 on basic surface centers. The catalytic activities of HT samples were different. The conversion of cyclohexanone increased from 30 to 37%, while the selectivity of ε-caprolactone decreased from 100 to 70% [15][40]. In addition, a series of tin-containing compounds of Sn-MCM-56 [16][41], Sn/HT [17][42], Sn-MCM-41 [18][43], Sn-palygorskite [19][44], P-PAMAM-HBA-Sn [20][45], Sn[N(SO2C8F17)2]4 [21][46], and Sn (salen)-NaY [22][47] were prepared. The catalytic activity tested in the BV reaction showed as being high. They are all heterogeneous catalysts and can be regenerated. Sn-MCM-56 was separated, washed with chlorobenzene, and calcined at 500 °C for 6 h. The catalytic activity of the regenerated catalyst was almost back to the initial level (conversion of 2-adamantanone: 40% (fresh), 38%(fifth)) (0.04 g Sn-MCM-56, n(adamantanone)/n(H2O2) = 2 mol/4 mol, 56 wt% H2O2, 4.5 g chlorobenzene, 90 °C, 2 h) [16][41]. Sn/HT was separated, and calcined at 400 °C for 2 h. The catalytic activity of the recycled Sn/HT was the same as that of the fresh one [17][42]. The catalytic activity of the regenerated P-PAMAM-HBA-Sn was lower than the fresh one (conversion of 2-adamantanone: 100% (fresh), 95 (second), 90 (third), 85 (fourth), 80% (fifth)) [20][45]. The catalytic activity of the regenerated Sn[N(SO2C8F17)2]4 was almost back to the initial level (conversion of 2-adamantanone: 93% (fresh), 92% (second), 90% (third), 90% (fourth)) [21][46]. Sn (salen)-NaY was separated by filtration and washed with 1,4-dioxane. The catalytic activity of the recycled Sn (salen)-NaY was the same as that of the fresh one (conversion of cyclohexanone: 75% (fresh), 74% (second), 75% (third)) [22][47]. The catalytic activity of Sn-bentonite formed by a simple procedure is low. It is necessary to add additives to improve the yield of lactones [23][48]. In addition, the toxicity of organotin compounds and tin chloride is high, and can induce DNA lesions. Although the toxicity of inorganic tin is low, its preparation uses tin chloride as a raw material [24][49]. Therefore new active, selective, and recyclable catalysts are still required, and a heterogeneous catalyst is an excellent option.
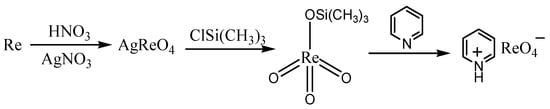
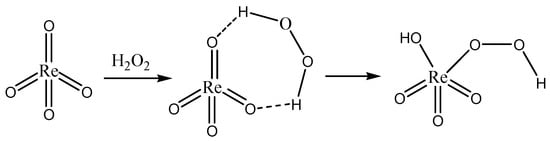
The synthesis of pyridinium perrhenate is shown in Figure 4 [25][50]. PyHReO4 was used to catalyze the BV oxidation of 2-adamantanone using 30% H2O2 as an oxidant, and the catalytic activity was high (yield: 90.02%; selectivity > 99%). PyHReO4 was separated by silica gel column chromatography. The catalytic activity of PyHReO4 in the oxidation of 2-adamantanone is attributed to the interaction between Re and hydrogen peroxide (Figure 5).
Silver supported on montmorillonite clay (Ag-NPs@mont) was synthesized and used as active catalysts in the BV oxidation of ketones with hydrogen peroxide as an oxidant under the solvent-free condition at room temperature [26][51]. The Ag-NPs@mont nanocatalyst was separated by filtration, washed with acetone, and dried. The recycled Ag-NPs@mont nanocatalyst was reused four times without significant loss of catalytic activity under the same conditions. The conversion of cyclohexanone was 99% under solvent-free conditions. The results in the oxidation of various ketones showed that the conversion of cyclic ketones was higher than aromatic ketones because aromatic ketones are more stable than cycloketones.
The magnetic composite Fe3O4@I-SR was prepared by loading Fe3O4 nanoparticles onto a porous illite silicon slag (I-SR) [27][52]. Fe3O4 nanoparticles were evenly distributed over the I-SR. Fe3O4@I-SR showed high catalytic activity in the oxidation of cyclohexanone. The conversion of cyclohexanone and the selectivity of ε-caprolactone were higher than 99%. Fe3O4@I-SR was separated, washed with acetone and ethanol, and dried. The catalytic activity of the recovered Fe3O4@I-SR was almost the same as that of the fresh one (conversion of cyclohexanone: 99% (fifth); selectivity of ε-caprolactone: 99% (fifth)). Fe3O4@I-SR can activate both the ketone group and H2O2, which can be used as catalysts to improve oxidation efficiency. Fe3O4@GO also showed high catalytic activity and selectivity for the BV oxidation of cyclohexanone without solvent [28][53].
Priya prepared mixed oxides of MoLaCeSm-Si [29][54]. MoLaCeSm-Si catalyst showed good activity and selectivity simultaneously in the oxidation of cyclohexanone to ε-caprolactone at room temperature. MoLaCeSm-Si was washed with acetone, dried, and activated at 400 °C for 1 h. The activity of the recovered MoLaCeSm-Si was not lost after six cycles. The conversion of cyclohexanone in the first cycle was 83.1%, and the selectivity of ε-caprolactone was 88.9%. The conversion of cyclohexanone was 79.1%, and the selectivity of ε-caprolactone was 81.2%, with the catalyst of MoLaCeSm-Si recovered from the sixth cycle. MoO3, La2O3, CeO2, and Sm2O3 in MoLaCeSm-Si each contribute their characteristic properties to increase the catalytic activity towards the oxidation of cyclohexanone at room temperature. Mo6+ in MoLaCeSm-Si provides the Lewis acidic center, [30][55] lanthanum induces primary sites on MoLaCeSm-Si [31][56], and ceriasamaria increases the oxygen vacancies [32][57] and performs the role of oxygen carrier between H2O2 and cyclohexanone.
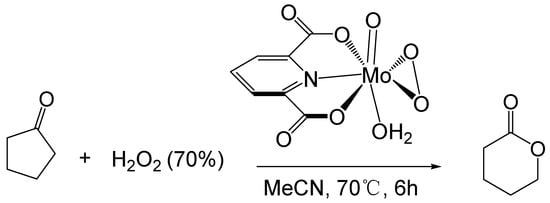
Furia [33][58] studied the BV reaction of ketones catalyzed by transition metals. They used a 70% hydrogen peroxide solution as an oxidant and a molybdenum peroxy complex as a catalyst to oxidize cyclopentanone to δ-valerolactone (Figure 6). The maximum conversion of cyclopentanone is 65%, and the maximum yield of δ-valerolactone is 54%. (0.09 M catalyst, 1.4 M cyclopentanone, 2.2 M H2O2, 70 °C, 6 h)
MoO3, WO3, and [{γ-SiW10O32(H2O)2}2(μ-O)2]4− have high catalytic activity and selectivity in the BV oxidation of cyclic ketones [34][35][59,60]. The catalytic activity of the recycled [{γ-SiW10O32(H2O)2}2(μ-O)2]4− was the same as that of the fresh one (yields of lactone: 99% (third run)). Cyclic ketones with tertiary carbon atoms adjacent to the ketone carbonyl group are more likely to undergo the BV reaction than cyclic ketones with secondary carbon atoms adjacent to the ketone carbonyl group, and the selectivity for the formation of corresponding lactones is also higher.
The high catalytic activity of MoO3 is due to its reaction with hydrogen peroxide to form peroxy acids. The possible reaction process is shown in Figure 7.
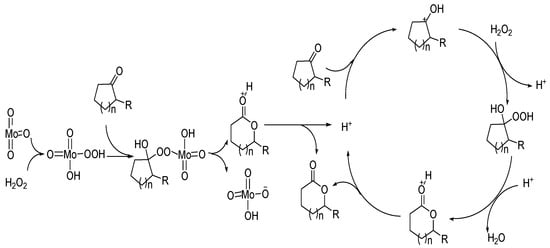
The catalytic mechanisms of Bronsted acids and Lewis acids are different. The good catalytic performance of transition metal oxides can be attributed to the coordination of the Lewis acid center of metal with carbonyl oxygen, and the reaction with hydrogen peroxide to form peroxy acids.布朗斯台德酸和路易斯酸的催化机制不同。过渡金属氧化物良好的催化性能可归因于金属的路易斯酸中心与羰基氧的配位,以及与过氧化氢反应生成过氧酸。