1. Noradrenergic Regulation of Spike-Wave Activity
To date, a large amount of data has been accumulated on the noradrenergic modulation of spike-wave activity (e.g., [1][2][3][4][40,41,42,43]). Cortico-thalamo-cortical neural circuitry is known to receive dense innervations from noradrenergic neurons [5][6][7][44,45,46]. Nevertheless, existing anti-absence drugs do not directly affect noradrenaline-related mechanisms or molecular targets of noradrenaline (NA). Mechanisms of noradrenergic regulation of arousal level may play a significant role in the generation of SWDs. By participating in state transitions, the NA system sets favorable conditions at molecular, cellular, and network levels for the appearance of pathological spike-wave activity. We suppose that by separating a contribution of the noradrenergic system into sleep-wake cycling and its modulation of spike-wave activity, novel, more selective pharmacological targets could be identified.
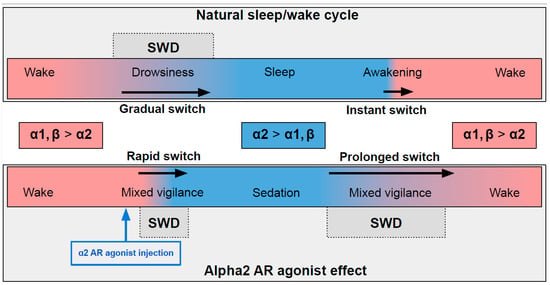
In naturally falling asleep, the NA level gradually decreases [8][80] and the predominantly alpha1- and beta-AR-mediated excitatory effect of NA switches to an alpha2-mediated inhibitory effect. This state is favorable for triggering spike-wave activity [9][10][28,77] (the upper part in Figure 1) Natural awakening provides an almost instant turn to arousal with a very low probability of SWDs occurring [9][11][28,81]. Injections of alpha2 ARs agonists might cause an unnaturally rapid increase in activation of alpha2 ARs leading to an artificial predominance of alpha2-mediated effect over the effect of other ARs subtypes (the bottom part of Figure 1). The emergence from sedation is again an extended transition favorable for spike-wave discharges (SWDs) to occur. Future studies of pharmacologically induced absence status epilepticus are required to examine the dose-dependent effects of alpha2 ARs agonists on SWDs. Another intriguing issue regarding the role of each ARs subtypes in the modulation of spike-wave activity and arousal level.
Figure 1. The schema demonstrating adrenergic mechanisms of sleep modulation and spike-wave discharge (SWD) modulation. The upper plot demonstrates the natural drug-free state. The bottom plot—pharmacologically induced condition after administration of agonist of alpha2 adrenoreceptors. Noradrenaline affects alpha1, alpha2 and beta-receptors, and NA concentration during sleep/sedation is lower (blue area) than during wakefulness (rose area). Transient states between wake and sleep/sedation are favorable for SWD to occur.
The influence of alpha2 ARs is mainly carried out through Gi/o-proteins although coupling to Gs was also demonstrated [12][103]. Thus, activation of alpha2 ARs can either inhibit or stimulate various intracellular pathways. Numerous neuronal proteins interacting directly or indirectly with alpha2 ARs have been described. Here we focus on phenomena related to spike-wave activity, such as interaction with ionic channels.
2. Alpha2-Adrenoreceptors and HCN Channels
HCN channels control neuron excitability and participate in the stabilization of resting potential
[13][104]. Due to their unique ability to open during hyperpolarization creating an inward current named I
h, activation of HCN channels may depolarize the neuron enough to initiate rebound bursting, a form of spiking activity crucial for spike-wave discharges
[14][105]. There are four HCN isoforms (HCN1–4). HCN1 isoform is mainly expressed in the cerebral cortex, and HCN2 is mostly in the thalamus but also in the cortex, and HCN4 in the thalamus (with the exception of the reticular nucleus) but not in the cortex. HCN3 is expressed at a low level in both structures and thus will not be analyzed further
[14][15][16][17][105,106,107,108]. HCN2 channels activate slower and at more negative voltages and have an increased sensitivity to cAMP modulation than HCN1
[18][19][109,110]. An increase in cAMP leads to a more pronounced shift in the activation of HCN2 channels into the depolarized direction, which leads to the elimination of differences in the degree of hyperpolarization needed to activate both isoforms. HCN4 has the slowest kinetics and is strongly modulated by cAMP
[20][21][111,112]. Melis Yavuz and Filiz Onat in 2018 published a comprehensive review about the role of HCN channels in the pathophysiology of absence epilepsy (also in rat models), where they emphasized the involvement of the second messenger system in modulating HCN channels
[22][113].
In different models of absence epilepsy and under different registration conditions, sometimes opposite changes in the HCN channels were found
[23][24][25][114,115,116]. However, it can be concluded that blocking the I
h current in the cortex leads to an increase in the excitability of neurons
[26][117]. This effect is based on enhanced temporal summation of the distal dendritic excitatory postsynaptic potentials, which can participate in SWDs generation
[27][28][118,119]. Conversely, blocking the I
h current in the thalamus suppresses burst firing and SWDs
[29][120]. HCN channels have an increasing distal expression gradient on dendrites (that is, the farther away from the soma, the denser)
[30][121], thus they provide signal filtering by weakening the contribution of remote excitatory postsynaptic potentials, EPSPs
[31][122].
The interaction of alpha2 ARs and HCN channels was studied in the context of epileptogensis
[31][32][122,123] and pain relief
[33][124]. It has been shown that activation of postsynaptic alpha2 ARs blocks I
h current, which increases input resistance and enhances temporal summation during trains of distally evoked EPSPs making a cell more excitable
[34][125]. Whether this mechanism is sufficient to aggravate spike-wave activity is yet to be investigated. Studies of the combined effects of the local application of substances acting on alpha2 ARs and HCN channels may clarify the role of their interaction in the spike-wave initiating site in the somatosensory cortex.
If alpha2 AR agonists inhibit HCN, then injection of alpha2 AR agonists into the thalamus should have an anti-absence effect, because blockage of the I
h current in thalamic neurons suppresses SWDs
[18][109]. This contradicts the results of Buzsáki et al. (1991)
[2][41]. Therefore, the effect of alpha2 AR agonists on I
h current may differ in the thalamus and in the cortex
[26][117]. Differences in the molecular cascades triggered by the activation of alpha2 AR subtypes are mostly studied in cell expression systems and need to be proved in vivo. Nevertheless, activation of the alpha2B subtype, predominantly expressed in the thalamus, may have a stimulatory effect on cAMP synthesis in contrast to the inhibitory action of alpha2A ARs
[35][126]. Selective coupling of alpha 2-adrenergic receptor subtypes to cyclic AMP-dependent reporter gene expression in transiently transfected JEG-3 cells.
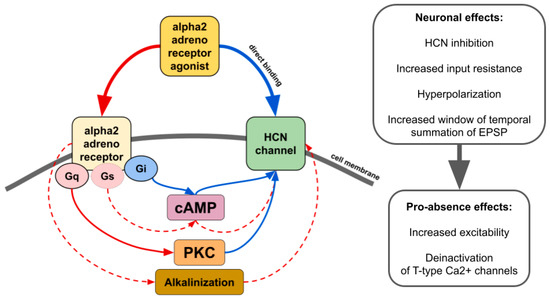
There is contradictory data on whether alpha2 AR agonists affect HCN channels through the modulation of cAMP synthesis or other pathways such as activation of protein kinase C
[4][43] or even by direct binding in an alpha2 AR-independent manner (
Figure 2)
[4][36][37][38][43,127,128,129]. The resulting effect seems to be dependent on which subtype of alpha2 and which isoforms of HCN interact. For example, increasing cAMP counteracts the inhibitory effect of ZD7288, a potent blocker of I
h, or dexmedetomidine and guanfacine on HCN channels
[37][39][40][128,130,131]. Thus, it may increase or even reverse inhibitory effects in tissues where cAMP-sensitive isoforms of HCN are highly expressed. Another way of alpha2-mediated upregulation of HCN channels is cellular alkalinization
[41][132] which shifts I
h activation potential to more depolarized values
[42][133]. A recent study showed that in rats genetically predisposed to absence epilepsy, spike-wave activity can be triggered by hypoxia-induced blood alkalosis resulting in the activation of neurons in the intralaminar thalamic nuclei
[43][134].
Figure 2. The main effects of alpha2 AR agonists on HCN channels. Red lines—activation. Blue lines—inhibition. Solid lines—effects have strong empirical evidence. Dashed lines—effects have limited evidence.
It should be mentioned that the impact of I
h on neuron excitability is intertwined with currents mediated by other ion channels such as G protein-coupled inwardly rectifying K+ channels
[44][135], which themselves may be influenced by alpha2 AR agonists. Dexmedetomidine activates a G protein-coupled inwardly rectifying K+ current
[45][136] which is suppressed by the first choice anti-absence drug ethosuximide
[46][137]. Considering the impending revision of the mechanism of action of ethosuximide as an anti-absence drug
[46][137], it is interesting to compare its effects on cell currents with the effects of alpha2 AR antagonists.
3. Alpha2-Adrenoreceptors and Calcium Channels
Calcium channels are important regulators of neuronal firing properties (reviewed in
[47][138]). Calcium channels are divided into low-voltage activated (T-type) and high-voltage activated (L-, N-, P/Q-, R-types). The role of calcium channels in the absence epilepsy is confirmed by genetic findings in patients and studies in knockout animal models. Pharmacological modulation of the spike-wave activity using calcium channel blockers and activators has been shown in animals genetically predisposed to absence seizures. In particular, intraperitoneal and intracerebroventricular microinjections of L-type blockers resulted in an increase in the number and duration of SWDs, but intracortical microinjections suppressed SWDs
[48][49][139,140].
Systemic administration of L-current activator BAY K8644 decreased the number and duration of spike-wave discharges, regardless of the method of administration. L-type calcium channel blockers are known to modulate the sedative effects of alpha-2 AR agonists. Local administration of nifedipine into LC and a subhypnotic dose of dexmedetomidine led to the loss of righting reflex
[50][141]; intraperitoneal injection of a low dose of nifedipine (2 mg/kg) blocked the sedative effect of clonidine
[51][142], and the high dose (20 mg/kg) enhanced dexmedetomidine-induced sleep time
[52][143]. Nevertheless, the sedative effects of alpha2 AR agonists were at least partially mediated by the blocking of L-type calcium channels, since the injection of nifedipine restored the hypnotic ability of dexmedetomidine in rats who developed tolerance after chronic administration
[52][143]. Noteworthy, the method of administration determined how an L-current blocker would affect a decrease in blood pressure induced by clonidine. Intravenous injection of nifedipine induced an increase in blood pressure in rabbits, but intravenous administration prevented the hypotensive action of clonidine
[53][144]. Since L-type blockers are used as antihypertensive drugs, their action on spike-wave activity and locomotion after systemic administration might be accounted for a decrease in blood pressure. Indeed, SWDs in humans and animals are known to be linked with a reduction in blood pressure
[54][145]. Identification of the contribution of L-current into pro-absence and sedative effects of alpha2 AR activation is possible with the local co-administration of substances into the central nervous system.
Alpha2 agonists reduce N- and P/Q-type currents
[12][55][56][103,146,147]. There is only limited evidence of the effects of N- or P/Q blockers on spike-wave activity, but they were either indistinguishable from the general detrimental influence of N-blockers or minor, as in the case of P/Q blockers
[48][139]. However, since mutations in high-voltage gated calcium channels lead to the development of absence epilepsy phenotypes in animal models
[57][58][148,149], the study of alpha2 ARs activation or inhibition in these models may indicate other ways to modulate spike-wave activity.
Altogether, T-type calcium channels play a crucial role in the generation of SWDs
[59][60][150,151], and interactions between alpha2 ARs and T-type calcium channels need to be further investigated. An increase in thalamic burst firing mediated by T-type channels might not be as significant in the generation of SWDs as previously assumed
[61][62][152,153]. Therefore, future studies are required to examine the relationship between the burst firing of thalamic cells and the SWD-promoting effects of alpha2 AR agonists.
4. Astrocytic Alpha2-Adrenoreceptors
More and more evidence has been obtained in favor of the fact that astrocytes can play an important role in the pathogenesis of epilepsy
[63][64][154,155]. Most studies have demonstrated the involvement of astrocytes in the pathogenesis of focal epilepsies
[63][64][65][66][154,155,156,157], but recently the role of astrocytes in the pathogenesis of absence epilepsy has been disclosed. Optogenetic excitation of astrocytes in the ventrobasal thalamus caused a pro-absence effect in GAERS and WAG/Rij rats
[67][158].
Astrocytes might be a promising target for the therapy of absence epilepsy. Alpha2A ARs are highly expressed in astrocytes, where they potentiate glutamate synthesis
[68][159] and stimulate GABA release
[69][160]. Although the general increase in GABA mediates the therapeutic effects of valproate, the administration of GABAa or GABAb agonists into the ventrobasal thalamus leads to an increase in spike-wave activity
[70][71][161,162]. Given the special role of astrocytes in controlling the level of GABA in the thalamus, where they provide the main contribution to the reuptake of this neurotransmitter
[72][163], they can be suggested as a target for novel drugs. It can be assumed that blocking thalamic alpha2 ARs with the selective antagonist to subtype B can reduce the level of GABA and prevent hyperpolarization of relay neurons, which creates a predisposition for burst firing.