Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 3 by Rita Xu.
C-reactive protein (CRP) is the final product of the interleukin (IL)-1β/IL-6/CRP axis. Its monomeric form can be produced at sites of local inflammation through the dissociation of pentameric CRP and, to some extent, local synthesis. Monomeric CRP (mCRP)mCRP has a distinct proinflammatory profile. In vitro and animal-model studies have suggested a role for mCRP in: platelet activation, adhesion, and aggregation; endothelial activation; leukocyte recruitment and polarization; foam-cell formation; and neovascularization. mCRP has been shown to deposit in atherosclerotic plaques and damaged tissues.
- monomeric C-reactive protein
- mCRP
- C-reactive protein
- biomarker
- atherosclerosis
- inflammation
1. Introduction
Atherosclerosis and its complications, primarily coronary artery disease (CAD) and ischemic stroke, remain the leading causes of mortality and disability worldwide. The incidence of atherosclerotic cardiovascular disease and major adverse cardiovascular events (MACE) increases with age. The probability of the clinical manifestation of atherosclerosis and the development of MACE is determined by the total plaque burden [1]. The total plaque burden is characterized by the concentration and duration of exposure to circulating atherogenic apolipoprotein B (apoB)-containing lipoproteins [1]. As plaque burden increases, the probability of atherosclerotic cardiovascular disease onset increases [1][2]. Therefore, reducing low-density lipoprotein cholesterol (LDL-C) level, the main fraction of apoB-containing lipoproteins in the blood, is the mainstay of atherosclerosis prevention. The reduction in LDL-C levels is achieved through diet, lifestyle modification, and pharmacological treatment with statin monotherapy, statin in combination with ezetimibe or proprotein convertase subtilisin-kexin type 9 (PCSK9) inhibitors [3].
Large randomized clinical trials demonstrated that aggressive lipid-lowering therapy with statins in combination with PCSK9 inhibitors produced a formidable reduction in LDL-C levels, approaching extremely low values in some patients. For example, a subgroup of 504 patients in the FOURIER trial achieved an LDL-C level of 0.18 mmol/L [4], whereas a subgroup of 3357 patients in the ODYSSEY OUTCOMES trial achieved an LDL-C level of less than 0.65 mmol/L [5]. Nevertheless, the MACE rate observed in these patients was substantial [6], which could be ascribed to the large total plaque burden that had already accumulated in these patients prior to treatment [1][2].
The cardiovascular risk that persists despite aggressive lipid-lowering therapy and correction of modifiable risk factors is called residual cardiovascular risk [7]. One of its main types is the residual inflammatory risk resulting from low-grade inflammation in atherosclerotic plaques [8]. It is determined by the level of the main inflammatory biomarker C-reactive protein (CRP), measured using a high-sensitivity assay (hsCRP), with a value of 2.0 mg/L or more [9]. The hsCRP assay measures the level of the pentameric form of CRP (pCRP), which is produced in the liver under the stimulation by interleukin (IL)-6 [10]. There exists another form of CRP, monomeric CRP (mCRP), which is formed at sites of local inflammation through the dissociation of pCRP and, to some extent, local synthesis. mCRP is essentially different from pCRP in its functions [10][11].
2. NLRP3 Inflammasome and Inflammatory IL-1β/IL-6/CRP Axis in Pathogenesis of Subclinical Vascular Inflammation
Cellular immune responses play a key role in all stages of atherosclerotic lesion development, from fatty streaks to plaque rupture [12]. A detailed discussion of cellular immunity in the pathogenesis of atherosclerosis has been provided elsewhere [13][14]. Four types of cells are involved in the pathogenesis of atherosclerotic lesions: smooth muscle cells (SMCs), T-lymphocytes, macrophages, and neutrophils. The cytoplasm of myeloid immune cells, including macrophages and neutrophils, and SMC contains pattern-recognition receptors (PRRs) [15][16]. These receptors recognize exogenous pathogen-associated molecular patterns (PAMPs) and endogenous damage-associated molecular patterns (DAMPs). DAMPs arise from signals produced by injured cells and tissues in the absence of an exogenous pathogen [16]. One type of PRR is the nucleotide-binding oligomerization domain-like receptor (NLR) [17]. The NLRP3 receptor in this group recognizes non-pathogenic DAMPs. NLRP3 activation leads to the formation of a molecular complex, the inflammasome, in the cytoplasm of myeloid immune cells. The NLRP3 inflammasome is a cytoplasmic protein complex that serves as a molecular platform for the activation of the cysteine protease caspase-1 [17]. Caspase-1 proteolyzes three proteins: pro-interleukin (pro-IL)-1β, pro-IL-18, and gasdermin D. Pro-IL-1β is converted into the active proinflammatory form of IL-1β and pro-IL-18 is converted into active proinflammatory IL-18. The activation of gasdermin D can induce pyroptosis, an inflammatory cell death, with the ensuing release of cytoplasmic content into surrounding tissues. This is particularly observed in the apoptosis of foam cells in atherosclerotic lesions [18][19]. The NLRP3 inflammasome formation has been observed in a number of diseases characterized by sterile inflammation. Uric-acid crystals can activate the NLRP3 inflammasome in gout [20]. Furthermore, the NLRP3 inflammasome plays an important role in the development of abdominal aortic aneurysm [21], myocardial damage in ischemia-reperfusion injury [22], kidney damage in diabetes, gout, and acute renal failure [23]. Active oxygen species and free fatty acids activate the NLRP3 inflammasome in obesity [24] and diabetes [25], contributing to the development of insulin resistance [26]. In 2010, Duewell et al. demonstrated for the first time the activation of the NLRP3 inflammasome by cholesterol crystals [27]. CD36-mediated uptake of oxidized LDL by macrophages results in intracellular cholesterol crystallization. This disrupts phagocytosis and leads to the accumulation of cholesterol crystals in macrophage lysosomes [28]. Subsequently, cholesterol crystals damage lysosome membranes and are released along with the lysosomal protease cathepsin B. They interact with PRR in the cytoplasm and induce the NLRP3 inflammasome formation [29]. Calcium-phosphate crystals accumulated in calcified atherosclerotic plaques can also activate the NLRP3 inflammasome [30]. Moreover, a recent study showed that CRP could activate the NLRP-3 inflammasome via the nuclear factor kappa B (NF-kB) pathway [31]. The NLRP3 inflammasome activation triggers a central cascade of inflammatory signaling represented by IL-1β, IL-6, and CRP [32]. IL-1β and IL-6 are mainly produced by myeloid cells [33][34], whereas CRP is by hepatocytes [10]. IL-1β stimulates the release of chemokines and the expression of cell-adhesion molecules by endotheliocytes, facilitating leukocyte recruitment to the site of inflammation. IL-1β stimulates the proliferation of SMC and the secretion of chemokines and collagenases by macrophages, thus contributing to the destabilization of atherosclerotic plaques [35]. IL-1β induces IL-6 synthesis. IL-6 has a wide range of proinflammatory and anti-inflammatory properties. For example, IL-6 stimulates the chemotaxis of neutrophils and macrophages to the site of inflammation [36]. It also stimulates chemokine release and expression of cell-adhesion molecules by endotheliocytes, facilitating leukocyte recruitment and platelet activation [37]. IL-6 produces a proatherogenic effect by stimulating SMC proliferation and modifying LDL uptake by macrophages and SMC [38]. Simultaneously, it stimulates an increase in LDL receptor expression, acting in an anti-atherogenic way [39]. IL-6 exhibits anti-inflammatory action by inhibiting IL-1 and tumor necrosis factor-α (TNF-α) synthesis and increasing IL-1 receptor antagonists and soluble p55 TNF-α receptor release [40]. Despite both proinflammatory and anti-inflammatory effects, higher levels of IL-1β and IL-6 are unequivocally associated with increased cardiovascular risk [41]. IL-6 stimulates CRP production in hepatocytes.3. Assessment of Residual Inflammatory Risk
3.1. C-Reactive Protein as a Biomarker of Subclinical Vascular Inflammation
CRP is the final product of the central inflammatory cascade. Owing to its excellent reproducibility, it is considered the main biomarker of inflammation that reflects the activity of the IL-1β/IL-6/CRP axis [32]. The CRP level is consistent over time. It is not affected by hematocrit or other blood-protein levels [42]. Moreover, it is unaffected by the circadian rhythm, time of food intake, blood sampling, anticoagulants, delay in specimen processing up to 6 h, or storage conditions. Nor is it affected by the specimen type: CRP measurements gave similar results in fresh, thawed, and even repeatedly thawed and refrozen plasma and serum. CRP can be measured using standard laboratory methods such as enzyme-linked immunosorbent assay (ELISA), turbidimetry, or nephelometry without significant discrepancies in results [42]. Thus, CRP is a convenient laboratory biomarker widely used in clinical practice and basic research. CRP levels rise manifold in response to infection or tissue damage: from 5–10 mg/L in mild cases to 320–550 mg/L in the most severe cases [43][44]. However, in atherosclerosis CRP levels are usually below 5 mg/L. A high-sensitivity assay with a threshold of 0.28 mg/L was developed to measure CRP below this level [45]. Large prospective observational studies have demonstrated that in surveyed populations, CRP levels were within tertiles of less than 1.0 mg/L, 1–3 mg/L, and more than 3.0 mg/L. In meta-analyses, the odds ratio for MACE between the lower and upper tertiles was between 1.58 and 2.0 [46][47]. The PROVE IT-TIMI 22 trial demonstrated that, in patients on aggressive statin therapy, the median CRP level was 2.0 mg/L. Patients with a CRP level of 2.0 mg/L or more had a 30% higher relative risk of MACE [48]. Similar CRP-level medians and cardiovascular risk ratios were observed in subsequent large clinical trials of statin therapy [49][50]. Currently, a CRP level 2.0 mg/L or more is suggested by the American College of Cardiology/American Heart Association guidelines on cardiovascular disease prevention as a cardiovascular risk factor [9]. The association between the CRP level and MACE rate has been examined in large observational studies in postmenopausal women [51], healthy volunteers in the Physicians’ Health Study [52], and MRFIT [53]. A meta-analysis of 52 prospective studies that included 246,669 individuals without cardiovascular disease showed that increased CRP levels worsened the 10-year prognosis of cardiovascular risk [54]. In addition, a meta-analysis of the East Asian population showed an association between elevated CRP and higher cardiovascular risk [55]. Furthermore, the USPSTF meta-analysis that explored studies published from 1966 to 2007 demonstrated that relative cardiovascular risk is 1.58-fold higher in individuals with a CRP level more than 3.0 mg/L than in those with a CRP level less than 1.0 mg/L [46].3.2. Pentameric C-Reactive Protein
pCRP belongs to the pentraxin family of acute-phase proteins. It is the primary acute-phase reactant in humans. pCRP is synthesized in hepatocytes and secreted into the bloodstream upon stimulation with IL-6. Circulating pCRP consists of five monomeric subunits bound with disulfide bonds in a ring-shaped disk [56]. Each subunit of the pentameric disk has a calcium-dependent binding site for lysophosphatidylcholine on one side and the complement component C1q on the other [57][58]. Phosphatidylcholine is a major structural component of cell and extracellular vesicle membranes. It is mainly present on the outer leaflet of the membrane phospholipid bilayer [59]. Secretory phospholipase A2 (PLA2) hydrolyzes it to lysophosphatidylcholine. Normally, phosphatidylcholine does not interact with PLA2. However, during apoptosis and cell injury, phospholipids of the inner leaflet translocate to the outer leaflet of the cell membrane. These phospholipids include phosphatidylserine and phosphatidylethanolamine, which are PLA2 ligands. In the presence of these two phospholipids, PLA2 hydrolyzes phosphatidylcholine to biologically active lysophosphatidylcholine [59]. In oxidized LDL, lipoprotein-associated PLA2 cleaves phosphatidylcholine in the lipid monolayer to lysophosphatidylcholine [60]. Lysophosphatidylcholine stimulates the endothelial synthesis of several chemokines, impairs endothelium-dependent arterial relaxation, increases oxidative stress, suppresses endotheliocyte migration and proliferation, and facilitates macrophage activation and polarization to the inflammatory M1 phenotype [61]. Lysophosphatidylcholine of oxidized LDL contributes to lysosomal damage and the NLRP-3 inflammasome activation in foam cells [62]. Lysophosphatidylcholine can activate the NLRP-3 inflammasome in adipose tissue, contributing to the development of insulin resistance [63]. Lysophosphatidylcholine is a ligand for pCRP [64]. Circulating pCRP acts as an opsonin that binds to lysophosphatidylcholine on the surface of cell membranes and oxidized lipoproteins. The sites on the reverse side of the CRP disc interact with the complement component C1q. This results in activation of the classical complement cascade up to the C4 component. Thus, CRP-induced complement activation facilitates phagocytosis of damaged cells and oxidized lipoproteins but does not initiate the formation of the membrane-attack complex C5b–C9 [64]. pCRP can also interact with factor H and activate the complement cascade up to the C4 component via the alternative pathway [65]. Furthermore, pCRP can opsonize nuclear antigens released by apoptotic and necrotic cells [66]. Therefore, the biological role of circulating pCRP is characterized by the facilitation of the clearance of cell-destruction products formed during trauma, infection, or sterile inflammation.3.3. Monomeric C-Reactive Protein
Upon binding to lysophosphatidylcholine, the pentameric disk of CRP undergoes dissociation through intermediate forms into the final product, mCRP [67][68]. Dissociation occurs through the disintegration of disulfide bonds between the pCRP subunits [69]. This process involves lysophosphatidylcholine but also requires other cell-membrane components and calcium. Soluble lysophosphatidylcholine does not dissociate pCRP in the absence of cell membranes [68]. Dissociation opens a neoepitope (octapeptide Phe-Thr-Lys-Pro-Gly-Leu-Trp-Pro) on the C-terminal end of monomeric subunits, which is concealed in the pentameric disk [68]. This dramatically changes the antigenic specificity and biological functions of CRP [70]. mCRP has reduced aqueous solubility and remains predominantly bound to the cell membranes [71]. It has been detected in extracellular vesicles circulating in the bloodstream. A pronounced increase in the number of mCRP-positive extracellular vesicles has been observed in patients with acute myocardial infarction [72] and peripheral artery disease [73]. mCRP in monocyte-derived exosomes has also been detected in patients with stable coronary artery disease [74]. mCRP may contribute to thromboinflammation. Thromboinflammation has recently been introduced to describe the complex interplay between blood coagulation and inflammation [75]. Immobilized on a collagen substrate, mCRP substantially increased platelet adhesion and thrombus growth rate at the shear rate of 1500 s−1, characteristic of arteries with mild stenosis [76]. Perfusion of mCRP-preincubated whole blood over a collagen type I-coated flow chamber yielded a similar result [76]. Unlike pCRP, mCRP induced platelet glycoprotein (GP) IIb/IIIa activation in a dose-dependent manner. mCRP facilitated platelet adhesion via activation of GP IIb/IIIa receptors. Moreover, pCRP dissociated into mCRP on activated adhered platelets during the perfusion of whole blood through a flow chamber [77]. Without dissociation, pCRP did not stimulate platelet adhesion or thrombus growth [76][77]. pCRP was attached to platelet membranes and dissociated into mCRP in another experiment with perfusion of whole blood over a surface coated with activated adhered platelets. GP IIb/IIIa inhibition with an antibody (abciximab) prevented pCRP dissociation [78]. mCRP stimulated platelet adhesion to the endothelial cells [79] and induced tissue-factor expression and fibrin formation on endothelial cells [80]. When dissociated on platelets and adhering to the vessel wall, mCRP can induce endothelial activation and leukocyte recruitment. mCRP enhanced endothelial activation and neutrophil attachment to the endothelium [79][81]; monocyte adhesion to the collagen [82], fibrinogen [83], and fibronectin matrix [84]; and T-lymphocyte extravasation [85]. In vitro, mCRP decreased nitric-oxide release and increased production of proinflammatory IL-8 and monocyte chemoattractant protein-1 by endothelial cells via the NF-kB pathway [86]. Moreover, mCRP stimulated leukocyte recruitment to the vessel wall, inducing the expression of vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin, as well as the production of IL-6 and IL-8 by the endothelium [79][86][87]. mCRP induced IL-8 production [71][88] and prevented neutrophil apoptosis [71]. In addition, mCRP stimulated macrophage and T-cell polarization to inflammatory M1 and Th1 phenotypes [89]. mCRP stimulated oxidized LDL uptake by macrophages [90]. The in vivo evidence that mCRP can stimulate monocyte infiltration into damaged tissues was obtained from recent studies on a murine model of myocardial infarction [91] and a rat model of renal ischemia/reperfusion injury [92]. Compared with controls, the infarcted myocardium of mCRP-pretreated mice demonstrated increased accumulation of macrophages of the inflammatory M1 phenotype [91]. Furthermore, the damaged renal tissue of pCRP-pretreated rats demonstrated increased infiltration with monocytes colocalized with mCRP [92]. In addition, mCRP has been shown to stimulate neoangiogenesis and stabilize novel microvessels in vitro (bovine aortic endothelial cells and SMC) and in vivo (chorioallantoic membrane) [93][94]. In summary, the role of mCRP has been suggested in: platelet activation, adhesion, and aggregation; endothelial activation; leukocyte recruitment and polarization; foam-cell formation; and neovascularization (Figure 1).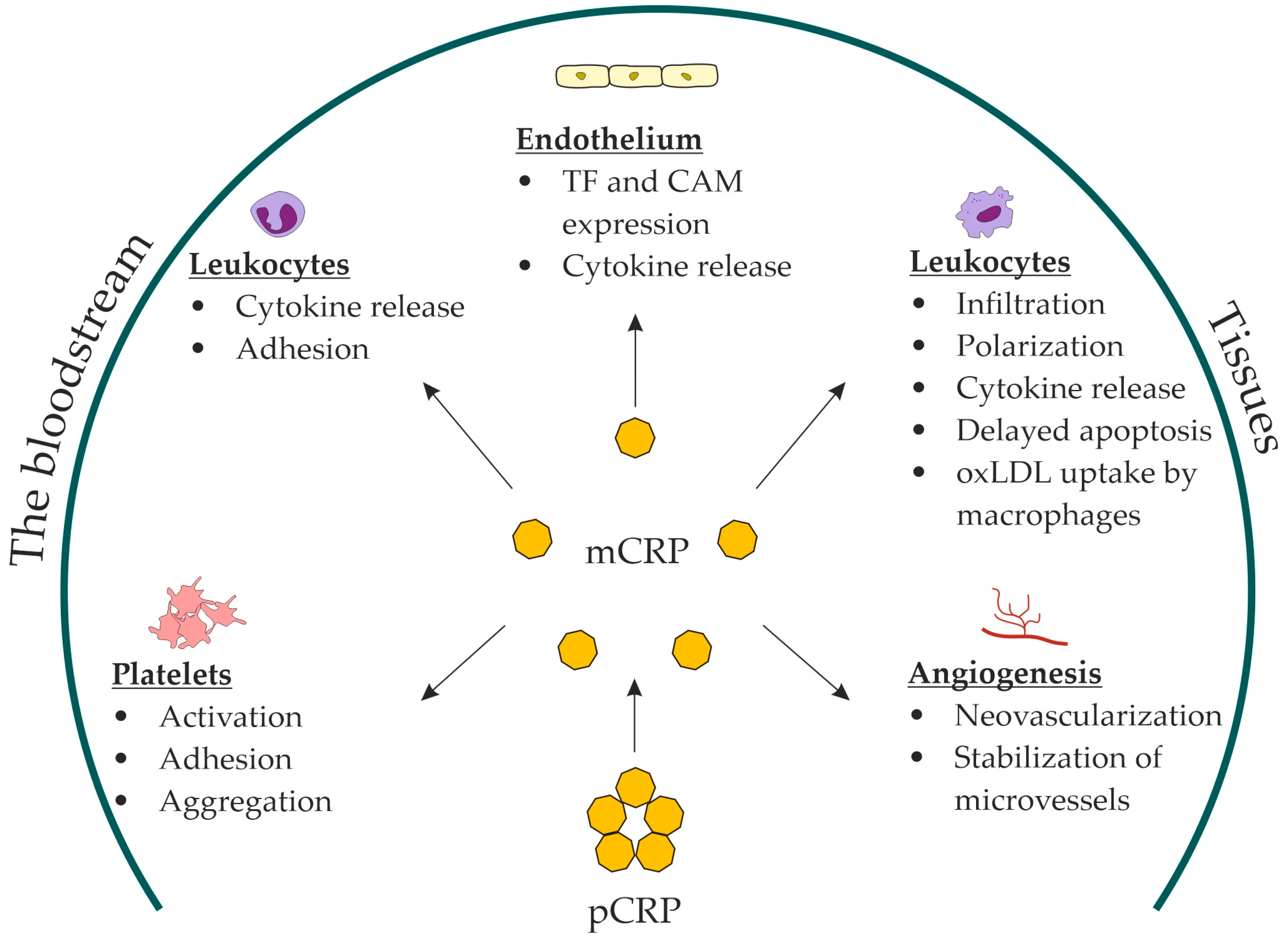
Figure 1. Proposed roles for mCRP in atherosclerosis. mCRP, monomeric C-reactive protein; pCRP, pentameric C-reactive protein; TF, tissue factor; CAM, cell-adhesion molecules; oxLDL, oxidized low-density lipoproteins.
References
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of Lipids on Cardiovascular Health. J. Am. Coll. Cardiol. 2018, 72, 1141–1156.
- Robinson, J.G.; Williams, K.J.; Gidding, S.; Borén, J.; Tabas, I.; Fisher, E.A.; Packard, C.; Pencina, M.; Fayad, Z.A.; Mani, V.; et al. Eradicating the Burden of Atherosclerotic Cardiovascular Disease by Lowering Apolipoprotein B Lipoproteins Earlier in Life. JAHA 2018, 7, e009778.
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188.
- Giugliano, R.P.; Pedersen, T.R.; Park, J.-G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971.
- Schwartz, G.G.; Gabriel Steg, P.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Kim, Y.-U.; Li, Q.H.; Manvelian, G.; et al. Clinical Efficacy and Safety of Alirocumab After Acute Coronary Syndrome According to Achieved Level of Low-Density Lipoprotein Cholesterol: A Propensity Score–Matched Analysis of the ODYSSEY OUTCOMES Trial. Circulation 2021, 143, 1109–1122.
- Dimmitt, S.B.; Stampfer, H.G.; Martin, J.H.; Warren, J.B. Clinical benefits of evolocumab appear less than hoped. Lancet 2018, 391, 933–934.
- Lawler, P.R.; Bhatt, D.L.; Godoy, L.C.; Lüscher, T.F.; Bonow, R.O.; Verma, S.; Ridker, P.M. Targeting cardiovascular inflammation: Next steps in clinical translation. Eur. Heart J. 2020, 42, ehaa099.
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur. Heart J. 2016, 37, 1720–1722.
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646.
- McFadyen, J.D.; Kiefer, J.; Braig, D.; Loseff-Silver, J.; Potempa, L.A.; Eisenhardt, S.U.; Peter, K. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol. 2018, 9, 1351.
- Rajab, I.M.; Hart, P.C.; Potempa, L.A. How C-Reactive Protein Structural Isoforms With Distinctive Bioactivities Affect Disease Progression. Front. Immunol. 2020, 11, 2126.
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126.
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610.
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327.
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340.
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Sig. Transduct. Target. Ther. 2021, 6, 291.
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451.
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Trans. Res. 2021, 14, 23–34.
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942.
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T.; et al. Inflammasome Activation by Mitochondrial Oxidative Stress in Macrophages Leads to the Development of Angiotensin II–Induced Aortic Aneurysm. ATVB 2015, 35, 127–136.
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604.
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity: Inflammasomes in kidney disease. Nephrology 2016, 21, 736–744.
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150.
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850.
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820.
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727.
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168.
- Bian, F.; Yang, X.-Y.; Xu, G.; Zheng, T.; Jin, S. CRP-Induced NLRP3 Inflammasome Activation Increases LDL Transcytosis Across Endothelial Cells. Front. Pharmacol. 2019, 10, 40.
- Ridker, P.M. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the Treatment of Atherothrombosis. Circ Res 2019, 124, 437–450.
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regener. 2019, 39, 12.
- Rose-John, S. Interleukin-6 signalling in health and disease. F1000Research 2020, 9, 1013.
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy. J. Am. Coll. Cardiol. 2017, 70, 2278–2289.
- Kaplanski, G. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29.
- Roldan, V. Interleukin-6, endothelial activation and thrombogenesis in chronic atrial fibrillation. Eur. Heart J. 2003, 24, 1373–1380.
- Morimoto, S.; Nabata, T.; Koh, E.; Shiraishi, T.; Fukuo, K.; Imanaka, S.; Kitano, S.; Miyashita, Y.; Ogihara, T. Interleukin-6 Stimulates Proliferation of Cultured Vascular Smooth Muscle Cells Independently of Interleukin-1β. J. Cardiovasc. Pharmacol. 1991, 17, S117–S118.
- Gierens, H.; Nauck, M.; Roth, M.; Schinker, R.; Schürmann, C.; Scharnagl, H.; Neuhaus, G.; Wieland, H.; März, W. Interleukin-6 Stimulates LDL Receptor Gene Expression via Activation of Sterol-Responsive and Sp1 Binding Elements. ATVB 2000, 20, 1777–1783.
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Res. 2011, 1813, 878–888.
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732.
- Aziz, N.; Fahey, J.L.; Detels, R.; Butch, A.W. Analytical Performance of a Highly Sensitive C-Reactive Protein-Based Immunoassay and the Effects of Laboratory Variables on Levels of Protein in Blood. Clin. Vaccine Immunol. 2003, 10, 652–657.
- Clyne, B.; Olshaker, J.S. The C-reactive protein. J. Emerg. Med. 1999, 17, 1019–1025.
- Póvoa, P.; Almeida, E.; Moreira, P.; Fernandes, A.; Mealha, R.; Aragão, A.; Sabino, H. C-reactive protein as an indicator of sepsis. Intensive Care Med. 1998, 24, 1052–1056.
- Eda, S.; Kaufmann, J.; Molwitz, M.; Vorberg, E. A new method of measuring C-reactive protein, with a low limit of detection, suitable for risk assessment of coronary heart disease. Scand. J. Clin. Lab Investig. Suppl. 1999, 230, 32–35.
- Buckley, D.I.; Fu, R.; Freeman, M.; Rogers, K.; Helfand, M. C-Reactive Protein as a Risk Factor for Coronary Heart Disease: A Systematic Review and Meta-analyses for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2009, 151, 483.
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O.; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L.; et al. Markers of Inflammation and Cardiovascular Disease: Application to Clinical and Public Health Practice: A Statement for Healthcare Professionals From the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511.
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-Reactive Protein Levels and Outcomes after Statin Therapy. N. Engl. J. Med. 2005, 352, 20–28.
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Crowe, T.; Sasiela, W.J.; Tsai, J.; Orazem, J.; Magorien, R.D.; O’Shaughnessy, C.; Ganz, P. Statin Therapy, LDL Cholesterol, C-Reactive Protein, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 29–38.
- Morrow, D.A.; de Lemos, J.A.; Sabatine, M.S.; Wiviott, S.D.; Blazing, M.A.; Shui, A.; Rifai, N.; Califf, R.M.; Braunwald, E. Clinical Relevance of C-Reactive Protein During Follow-Up of Patients With Acute Coronary Syndromes in the Aggrastat-to-Zocor Trial. Circulation 2006, 114, 281–288.
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843.
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, Aspirin, and the Risk of Cardiovascular Disease in Apparently Healthy Men. N. Engl. J. Med. 1997, 336, 973–979.
- MRFIT Research Group; Kuller, L.H.; Tracy, R.P.; Shaten, J.; Meilahn, E.N. Relation of C-Reactive Protein and Coronary Heart Disease in the MRFIT Nested Case-Control Study. Am. J. Epidemiol. 1996, 144, 537–547.
- The Emerging Risk Factors Collaboration C-Reactive Protein, Fibrinogen, and Cardiovascular Disease Prediction. N. Engl. J. Med. 2012, 367, 1310–1320.
- Saito, I.; Maruyama, K.; Eguchi, E. C-Reactive Protein and Cardiovascular Disease in East Asians: A Systematic Review. Clin. Med. Insights Cardiol. 2014, 8, CMC-S17066.
- Lv, J.-M.; Lü, S.-Q.; Liu, Z.-P.; Zhang, J.; Gao, B.-X.; Yao, Z.-Y.; Wu, Y.-X.; Potempa, L.A.; Ji, S.-R.; Long, M.; et al. Conformational folding and disulfide bonding drive distinct stages of protein structure formation. Sci. Rep. 2018, 8, 1494.
- Volanakis, J.E.; Wirtz, K.W.A. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature 1979, 281, 155–157.
- Thompson, D.; Pepys, M.B.; Wood, S.P. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 1999, 7, 169–177.
- Hack, C.E.; Wolbink, G.-J.; Schalkwijk, C.; Speijer, H.; Hermens, W.T.; van den Bosch, H. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol. Today 1997, 18, 111–115.
- Wu, R.; Huang, Y.H.; Elinder, L.S.; Frostegård, J. Lysophosphatidylcholine Is Involved in the Antigenicity of Oxidized LDL. ATVB 1998, 18, 626–630.
- Law, S.-H.; Chan, M.-L.; Marathe, G.K.; Parveen, F.; Chen, C.-H.; Ke, L.-Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149.
- Corrêa, R.; Silva, L.F.F.; Ribeiro, D.J.S.; das Neves Almeida, R.; de Oliveira Santos, I.; Corrêa, L.H.; de Sant’Ana, L.P.; Assunção, L.S.; Bozza, P.T.; Magalhães, K.G. Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front. Immunol. 2020, 10, 2927.
- Zhang, S.-Y.; Dong, Y.-Q.; Wang, P.; Zhang, X.; Yan, Y.; Sun, L.; Liu, B.; Zhang, D.; Zhang, H.; Liu, H.; et al. Adipocyte-derived Lysophosphatidylcholine Activates Adipocyte and Adipose Tissue Macrophage Nod-Like Receptor Protein 3 Inflammasomes Mediating Homocysteine-Induced Insulin Resistance. EBioMedicine 2018, 31, 202–216.
- Du Clos, T.W.; Mold, C. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcγ receptors. Curr. Opin. Organ Transplant. 2011, 16, 15–20.
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30.
- Du Clos, T.W. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol. Biol. Rep. 1996, 23, 253–260.
- Braig, D.; Nero, T.L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188.
- Ji, S.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRPm. FASEB J. 2007, 21, 284–294.
- Zhang, C.-M.; Tan, Y.-B.; Zhou, H.-H.; Ge, Z.-B.; Feng, J.-R.; Lv, G.-B.; Sun, Z.-Y.; Fu, Y.; Wang, M.-Y. Intra-subunit Disulfide Determines the Conversion and Structural Stability of CRP Isoforms. Inflammation 2020, 43, 466–477.
- Li, H.-Y.; Wang, J.; Meng, F.; Jia, Z.-K.; Su, Y.; Bai, Q.-F.; Lv, L.-L.; Ma, F.-R.; Potempa, L.A.; Yan, Y.-B.; et al. An Intrinsically Disordered Motif Mediates Diverse Actions of Monomeric C-reactive Protein. J. Biol. Chem. 2016, 291, 8795–8804.
- Khreiss, T.; József, L.; Hossain, S.; Chan, J.S.D.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry of C-reactive Protein Is Associated with Delayed Apoptosis of Human Neutrophils. J. Biol. Chem. 2002, 277, 40775–40781.
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.-M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 2012, 96, 64–72.
- Crawford, J.R.; Trial, J.; Nambi, V.; Hoogeveen, R.C.; Taffet, G.E.; Entman, M.L. Plasma Levels of Endothelial Microparticles Bearing Monomeric C-reactive Protein are Increased in Peripheral Artery Disease. J. Cardiovasc. Trans. Res. 2016, 9, 184–193.
- Melnikov, I.; Kozlov, S.; Saburova, O.; Zubkova, E.; Guseva, O.; Domogatsky, S.; Arefieva, T.; Radyukhina, N.; Zvereva, M.; Avtaeva, Y.; et al. CRP Is Transported by Monocytes and Monocyte-Derived Exosomes in the Blood of Patients with Coronary Artery Disease. Biomedicines 2020, 8, 435.
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; ten Cate-Hoek, A.J.; ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564.
- Molins, B.; Peña, E.; Vilahur, G.; Mendieta, C.; Slevin, M.; Badimon, L. C-Reactive Protein Isoforms Differ in Their Effects on Thrombus Growth. ATVB 2008, 28, 2239–2246.
- Molins, B.; Peña, E.; de la Torre, R.; Badimon, L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 2011, 92, 328–337.
- de la Torre, R.; Peña, E.; Vilahur, G.; Slevin, M.; Badimon, L. Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: Pentraxins and platelet deposition. J. Thromb. Haemost. 2013, 11, 2048–2058.
- Khreiss, T.; József, L.; Potempa, L.A.; Filep, J.G. Conformational Rearrangement in C-Reactive Protein Is Required for Proinflammatory Actions on Human Endothelial Cells. Circulation 2004, 109, 2016–2022.
- Li, R.; Ren, M.; Luo, M.; Chen, N.; Zhang, Z.; Luo, B.; Wu, J. Monomeric C-reactive protein alters fibrin clot properties on endothelial cells. Thromb. Res. 2012, 129, e251–e256.
- Zouki, C.; Haas, B.; Chan, J.S.D.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry of C-Reactive Protein Is Associated with Promotion of Neutrophil-Endothelial Cell Adhesion. J. Immunol. 2001, 167, 5355–5361.
- Eisenhardt, S.U.; Habersberger, J.; Murphy, A.; Chen, Y.-C.; Woollard, K.J.; Bassler, N.; Qian, H.; von zur Muhlen, C.; Hagemeyer, C.E.; Ahrens, I.; et al. Dissociation of Pentameric to Monomeric C-Reactive Protein on Activated Platelets Localizes Inflammation to Atherosclerotic Plaques. Circ. Res. 2009, 105, 128–137.
- Thiele, J.R.; Habersberger, J.; Braig, D.; Schmidt, Y.; Goerendt, K.; Maurer, V.; Bannasch, H.; Scheichl, A.; Woollard, K.J.; von Dobschütz, E.; et al. Dissociation of Pentameric to Monomeric C-Reactive Protein Localizes and Aggravates Inflammation: In Vivo Proof of a Powerful Proinflammatory Mechanism and a New Anti-Inflammatory Strategy. Circulation 2014, 130, 35–50.
- Ullah, N.; Ma, F.-R.; Han, J.; Liu, X.-L.; Fu, Y.; Liu, Y.-T.; Liang, Y.-L.; Ouyang, H.; Li, H.-Y. Monomeric C-reactive protein regulates fibronectin mediated monocyte adhesion. Mol. Immunol. 2020, 117, 122–130.
- Zhang, Z.; Na, H.; Gan, Q.; Tao, Q.; Alekseyev, Y.; Hu, J.; Yan, Z.; Yang, J.B.; Tian, H.; Zhu, S.; et al. Monomeric C-reactive protein via endothelial CD31 for neurovascular inflammation in an ApoE genotype-dependent pattern: A risk factor for Alzheimer’s disease? Aging Cell 2021, 20, e13501.
- Li, H.-Y.; Wang, J.; Wu, Y.-X.; Zhang, L.; Liu, Z.-P.; Filep, J.G.; Potempa, L.A.; Wu, Y.; Ji, S.-R. Topological Localization of Monomeric C-reactive Protein Determines Proinflammatory Endothelial Cell Responses. J. Biol. Chem. 2014, 289, 14283–14290.
- Ji, S.-R.; Ma, L.; Bai, C.-J.; Shi, J.-M.; Li, H.-Y.; Potempa, L.A.; Filep, J.G.; Zhao, J.; Wu, Y. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 2009, 23, 1806–1816.
- Khreiss, T.; József, L.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry in C-Reactive Protein Induces Interleukin-8 Secretion Through Peroxynitrite Signaling in Human Neutrophils. Circ. Res. 2005, 97, 690–697.
- Trial, J.; Potempa, L.A.; Entman, M.L. The role of C-reactive protein in innate and acquired inflammation: New perspectives. Inflamm. Cell Signal. 2016, 3, e1409.
- Ji, S.; Wu, Y.; Potempa, L.; Qiu, Q.; Zhao, J. Interactions of C-reactive protein with low-density lipoproteins: Implications for an active role of modified C-reactive protein in atherosclerosis. Int. J. Biochem. Cell Biol. 2006, 38, 648–661.
- Zha, Z.; Cheng, Y.; Cao, L.; Qian, Y.; Liu, X.; Guo, Y.; Wang, J. Monomeric CRP Aggravates Myocardial Injury After Myocardial Infarction by Polarizing the Macrophage to Pro-Inflammatory Phenotype Through JNK Signaling Pathway. JIR 2021, 14, 7053–7064.
- Thiele, J.R.; Zeller, J.; Kiefer, J.; Braig, D.; Kreuzaler, S.; Lenz, Y.; Potempa, L.A.; Grahammer, F.; Huber, T.B.; Huber-Lang, M.; et al. A Conformational Change in C-Reactive Protein Enhances Leukocyte Recruitment and Reactive Oxygen Species Generation in Ischemia/Reperfusion Injury. Front. Immunol. 2018, 9, 675.
- Boras, E.; Slevin, M.; Alexander, M.Y.; Aljohi, A.; Gilmore, W.; Ashworth, J.; Krupinski, J.; Potempa, L.A.; Al Abdulkareem, I.; Elobeid, A.; et al. Monomeric C-reactive protein and Notch-3 co-operatively increase angiogenesis through PI3K signalling pathway. Cytokine 2014, 69, 165–179.
- Turu, M.M.; Slevin, M.; Matou, S.; West, D.; Rodríguez, C.; Luque, A.; Grau-Olivares, M.; Badimon, L.; Martinez-Gonzalez, J.; Krupinski, J. C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signalling pathways and gene expression. BMC Cell Biol. 2008, 9, 47.
- Krupinski, J.; Turu, M.M.; Martinez-Gonzalez, J.; Carvajal, A.; Juan-Babot, J.O.; Iborra, E.; Slevin, M.; Rubio, F.; Badimon, L. Endogenous Expression of C-Reactive Protein Is Increased in Active (Ulcerated Noncomplicated) Human Carotid Artery Plaques. Stroke 2006, 37, 1200–1204.
- Kobayashi, S.; Inoue, N.; Ohashi, Y.; Terashima, M.; Matsui, K.; Mori, T.; Fujita, H.; Awano, K.; Kobayashi, K.; Azumi, H.; et al. Interaction of Oxidative Stress and Inflammatory Response in Coronary Plaque Instability: Important Role of C-Reactive Protein. ATVB 2003, 23, 1398–1404.
- Melnikov, I.S.; Kozlov, S.G.; Chumachenko, P.V.; Saburova, O.S.; Guseva, O.A.; Prokofyeva, L.V.; Gabbasov, Z.A. Monomeric C-reactive protein and local inflammatory reaction in the wall of the coronary arteries in patients with stable coronary artery disease. Russ. J. Cardiol. 2019, 24, 56–61.
- Vainas, T.; Stassen, F.R.M.; de Graaf, R.; Twiss, E.L.L.; Herngreen, S.B.; Welten, R.J.T.J.; van den Akker, L.H.J.M.; van Dieijen-Visser, M.P.; Bruggeman, C.A.; Kitslaar, P.J.E.H.M. C-reactive protein in peripheral arterial disease: Relation to severity of the disease and to future cardiovascular events. J. Vasc. Surg. 2005, 42, 243–251.
- Jabs, W.J.; Theissing, E.; Nitschke, M.; Bechtel, J.F.M.; Duchrow, M.; Mohamed, S.; Jahrbeck, B.; Sievers, H.-H.; Steinhoff, J.; Bartels, C. Local Generation of C-Reactive Protein in Diseased Coronary Artery Venous Bypass Grafts and Normal Vascular Tissue. Circulation 2003, 108, 1428–1431.
- Sattler, K.J.E.; Woodrum, J.E.; Galili, O.; Olson, M.; Samee, S.; Meyer, F.B.; Zhu, X.-Y.; Lerman, L.O.; Lerman, A. Concurrent Treatment With Renin-Angiotensin System Blockers and Acetylsalicylic Acid Reduces Nuclear Factor κB Activation and C-Reactive Protein Expression in Human Carotid Artery Plaques. Stroke 2005, 36, 14–20.
- Calabro, P.; Chang, D.W.; Willerson, J.T.; Yeh, E.T.H. Release of C-Reactive Protein in Response to Inflammatory Cytokines by Human Adipocytes: Linking Obesity to Vascular Inflammation. J. Am. Coll. Cardiol. 2005, 46, 1112–1113.
- Kolb-Bachofen, V.; Puchta-Teudt, N.; Egenhofer, C. Expression of membrane-associated C-reactive protein by human monocytes: Indications for a selectin-like activity participating in adhesion. Glycoconj. J. 1995, 12, 122–127.
- Ciubotaru, I.; Potempa, L.A.; Wander, R.C. Production of Modified C-Reactive Protein in U937-Derived Macrophages. Exp. Biol. Med. 2005, 230, 762–770.
- Haider, D.G.; Leuchten, N.; Schaller, G.; Gouya, G.; Kolodjaschna, J.; Schmetterer, L.; Kapiotis, S.; Wolzt, M. C-reactive protein is expressed and secreted by peripheral blood mononuclear cells. Clin. Exp. Immunol. 2006, 146, 533–539.
More