Since the 1990s, ionotropic glutamate receptors have served as an outstanding target for drug discovery research aimed at the discovery of new neurotherapeutic agents. With the recent approval of perampanel, the first marketed non-competitive antagonist of AMPA receptors, particular interest has been directed toward ‘non-NMDA’ (AMPA and kainate) receptor inhibitors. Although the role of AMPA receptors in the development of neurological or psychiatric disorders has been well recognized and characterized, progress in understanding the function of kainate receptors (KARs) has been hampered, mainly due to the lack of specific and selective pharmacological tools. The latest findings in the biology of KA receptors indicate that they are involved in neurophysiological activity and play an important role in both health and disease, including conditions such as anxiety, schizophrenia, epilepsy, neuropathic pain, and migraine.
- glutamate ionotropic receptors
- kainate receptors
- antagonist
1. Competitive Antagonists of the Kainate Receptors
1.1. Quinoxaline-2,3-diones
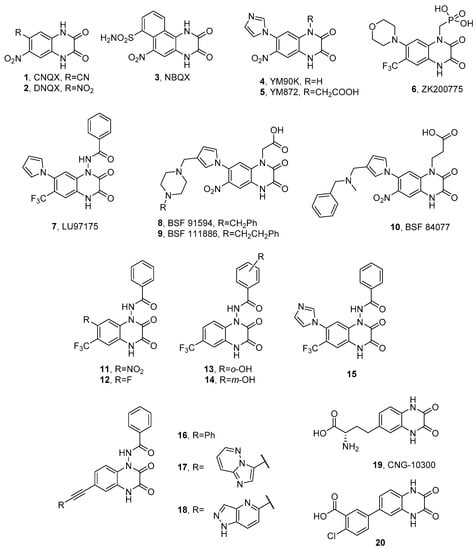
1.2. α-Amino Acid Antagonists
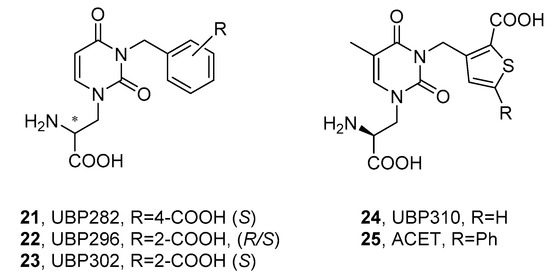
1.2.1. Willardiines
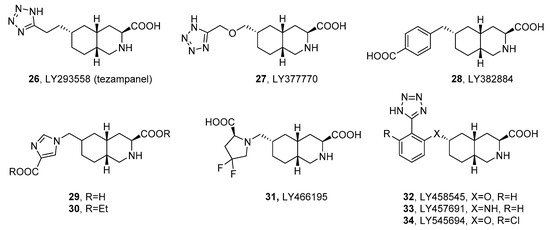
1.2. Decahydroisoquinolines
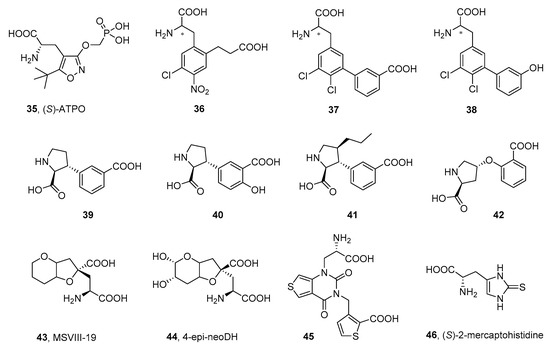
1.3. Other α-Amino Acid Antagonists
1.34. Structurally Dissimilar AMPAR/KAR Antagonists
2. Non-Competitive Antagonists and Channel Blockers of Kainate Receptors
References
- Honoré, T.; Davies, S.N.; Drejer, J.; Fletcher, E.J.; Jacobsen, P.; Lodge, D.; Nielsen, F.E. Quinoxalinediones: Potent competitive non-NMDA glutamate receptor antagonists. Science 1988, 241, 701–703.
- Sheardown, M.J.; Nielsen, E.O.; Hansen, A.J.; Jacobsen, P.; Honore, T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: A neuroprotectant for cerebral ischemia. Science 1990, 247, 571–574.
- Judge, M.E.; Sheardown, M.J.; Jacobsen, P.; Honoré, T. Protection against post-ischemic behavioral pathology by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) in the gerbil. Neurosci. Lett. 1991, 133, 291–294.
- Lubisch, W.; Behl, B.; Hofmann, H.P. Amido-Quinoxalinediones, the Preparation and Use Thereof. U.S. Patent 5,773,439, 30 June 1998.
- Löscher, W.; Lehmann, H.; Behl, B.; Seemann, D.; Teschendorf, H.J.; Hofmann, H.P.; Lubisch, W.; Höger, T.; Lemaire, H.G.; Gross, G. A new pyrrolyl-quinoxalinedione series of non-NMDA glutamate receptor antagonists: Pharmacological characterization and comparison with NBQX and valproate in the kindling model of epilepsy. Eur. J. Neurosci. 1999, 11, 250–262.
- Twele, F.; Bankstahl, M.; Klein, S.; Römermann, K.; Löscher, W. The AMPA receptor antagonist NBQX exerts anti-seizure but not antiepileptogenic effects in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Neuropharmacology 2015, 95, 234–242.
- Lippman-Bell, J.J.; Rakhade, S.N.; Klein, P.M.; Obeid, M.; Jackson, M.C.; Joseph, A.; Jensen, F.E. AMPA Receptor antagonist NBQX attenuates later-life epileptic seizures and autistic-like social deficits following neonatal seizures. Epilepsia 2013, 54, 1922–1932.
- Kong, L.-L.; Yu, L.-C. It is AMPA receptor, not kainate receptor, that contributes to the NBQX-induced antinociception in the spinal cord of rats. Brain Res. 2006, 1100, 73–77.
- Van Damme, P.; Leyssen, M.; Callewaert, G.; Robberecht, W.; Van Den Bosch, L. The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2003, 343, 81–84.
- Goda, M.; Isono, M.; Fujiki, M.; Kobayashi, H. Both MK801 and NBQX reduce the neuronal damage after impact-acceleration brain injury. J. Neurotrauma 2002, 19, 1445–1456.
- Follett, P.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 2000, 20, 9235–9241.
- Swedberg, M.D.; Jacobsen, P.; Honoré, T. Anticonvulsant, anxiolytic and discriminative effects of the AMPA antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX). J. Pharmacol. Exp. Ther. 1995, 274, 1113–1121.
- Matsuoka, Y.; Kitamura, Y.; Tsukahara, T.; Terai, K.; Tooyama, I.; Kimura, H.; Taniguchi, T. Neuroprotective effects of NBQX on hypoxia-induced neuronal damage in rat hippocampus. Neuroreport 1995, 6, 2205–2208.
- Löscher, W.; Hönack, D. Effects of the non-NMDA antagonists NBQX and the 2,3-benzodiazepine GYKI 52466 on different seizure types in mice: Comparison with diazepam and interactions with flumazenil. Br. J. Pharmacol. 1994, 113, 1349–1357.
- Catarzi, D.; Colotta, V.; Varano, F. Competitive AMPA receptor antagonists. Med. Res. Rev. 2007, 27, 239–278.
- Nikam, S.S.; Kornberg, B.E. AMPA receptor antagonists. Curr. Med. Chem. 2001, 8, 155–170.
- Larsen, A.M.; Bunch, L. Medicinal chemistry of competitive kainate receptor antagonists. ACS Chem. Neurosci. 2011, 2, 60–74.
- Shimizu-Sasamata, M.; Kawasaki-Yatsugi, S.; Okada, M.; Sakamoto, S.; Yatsugi, S.; Togami, J.; Hatanaka, K.; Ohmori, J.; Koshiya, K.; Usuda, S.; et al. YM90K: Pharmacological characterization as a selective and potent alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor antagonist. J. Pharmacol. Exp. Ther. 1996, 276, 84–92.
- Takahashi, M.; Kohara, A.; Shishikura, J.; Kawasaki-Yatsugi, S.; Ni, J.W.; Yatsugi, S.; Sakamoto, S.; Okada, M.; Shimizu-Sasamata, M.; Yamaguchi, T. YM872: A selective, potent and highly water-soluble alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor antagonist. CNS Drug Rev. 2002, 8, 337–352.
- Furukawa, T.; Hoshino, S.; Kobayashi, S.; Asakura, T.; Takahashi, M.; Atsumi, T.; Teramoto, A. The glutamate AMPA receptor antagonist, YM872, attenuates cortical tissue loss, regional cerebral edema, and neurological motor deficits after experimental brain injury in rats. J. Neurotrauma 2003, 20, 269–278.
- Turski, L.; Huth, A.; Sheardown, M.J.; McDonald, F.; Neuhaus, R.; Schneider, H.H.; Dirnagl, U.; Wiegand, F.; Jacobsen, P.; Ottow, E. ZK200775: A phosphonate quinoxalinedione AMPA antagonist for neuroprotection in stroke and trauma. Proc. Natl. Acad. Sci. USA 1998, 95, 10960–10965.
- Terai, K.; Suzuki, M.; Sasamata, M. Therapeutic Agent for Brain Hemorrhage. WO2004002488, 8 January 2004.
- Ishiuchi, S. Remedy for Glioblastoma. CA2479495, 9 October 2003.
- Pallesen, J.; Møllerud, S.; Frydenvang, K.; Pickering, D.S.; Bornholdt, J.; Nielsen, B.; Pasini, D.; Han, L.; Marconi, L.; Kastrup, J.S.; et al. N1-substituted quinoxaline-2,3-diones as kainate receptor antagonists: X-ray crystallography, structure−affinity relationships, and in vitro pharmacology. ACS Chem. Neurosci. 2019, 10, 1841–1853.
- Madsen, U.; Stensbol, T.; Krogsgaard-Larsen, P. Inhibitors of ampa and kainate receptors. Curr. Med. Chem. 2012, 8, 1291–1301.
- Lubisch, W.; Behl, B.; Henn, C.; Hofmann, H.P.; Reeb, J.; Regner, F.; Vierling, M. Pyrrolylquinoxalinediones carrying a piperazine residue represent highly potent and selective ligands to the homomeric kainate receptor GluR5. Bioorg. Med. Chem. Lett. 2002, 12, 2113–2116.
- Møllerud, S.; Hansen, R.B.; Pallesen, J.; Temperini, P.; Pasini, D.; Bornholt, J.; Nielsen, B.; Mamedova, E.; Chalupnik, P.; Paternain, A.V.; et al. N-(7-(1H-Imidazol-1-yl)-2,3-dioxo-6-(trifluoromethyl)-3,4-dihydroquinoxalin-1(2H)-yl)benzamide, a new kainate receptor selective antagonist and analgesic: Synthesis, X-ray crystallography, structure-affinity relationships, and in vitro and in vivo pharmacology. ACS Chem. Neurosci. 2019, 10, 4685–4695.
- Møllerud, S.; Frydenvang, K.; Pickering, D.S.; Kastrup, J.S. Lessons from crystal structures of kainate receptors. Neuropharmacology 2017, 112, 16–28.
- Pøhlsgaard, J.; Frydenvang, K.; Madsen, U.; Kastrup, J.S. Lessons from more than 80 structures of the GluA2 ligand-binding domain in complex with agonists, antagonists and allosteric modulators. Neuropharmacology 2011, 60, 135–150.
- Chałupnik, P.; Vialko, A.; Pickering, D.S.; Hinkkanen, M.; Donbosco, S.; Møller, T.C.; Jensen, A.A.; Nielsen, B.; Bay, Y.; Kristensen, A.S.; et al. Discovery of the first highly selective antagonist of the GluK3 kainate receptor subtype. Int. J. Mol. Sci. 2022, 23, 8797.
- Demmer, C.S.; Møller, C.; Brown, P.M.; Han, L.; Pickering, D.S.; Nielsen, B.; Bowie, D.; Frydenvang, K.; Kastrup, J.S.; Bunch, L. Binding mode of an α-amino acid-linked quinoxaline-2,3-dione analogue at glutamate receptor subtype GluK1. ACS Chem. Neurosci. 2015, 6, 845–854.
- Demmer, C.S.; Rombach, D.; Liu, N.; Nielsen, B.; Pickering, D.S.; Bunch, L. Revisiting the quinoxalinedione scaffold in the construction of new ligands for the ionotropic glutamate receptors. ACS Chem. Neurosci. 2017, 8, 2477–2495.
- More, J.C.A.; Troop, H.M.; Dolman, N.P.; Jane, D.E. Structural requirements for novel willardiine derivatives acting as AMPA and kainate receptor antagonists. Br. J. Pharmacol. 2003, 138, 1093–1100.
- More, J.C.A.; Nistico, R.; Dolman, N.P.; Clarke, V.R.J.; Alt, A.J.; Ogden, A.M.; Buelens, F.P.; Troop, H.M.; Kelland, E.E.; Pilato, F.; et al. Characterisation of UBP296: A novel, potent and selective kainate receptor antagonist. Neuropharmacology 2004, 47, 46–64.
- Mayer, M.L.; Ghosal, A.; Dolman, N.P.; Jane, D.E. Crystal structures of the kainate receptor GluR5 ligand binding core dimer with novel GluR5-selective antagonists. J. Neurosci. 2006, 26, 2852–2861.
- Dolman, N.P.; More, J.C.; Alt, A.; Knauss, J.L.; Pentikainen, O.T.; Glasser, C.R.; Bleakman, D.; Mayer, M.L.; Collingridge, G.L.; Jane, D.E. Synthesis and pharmacological characterization of N3-substituted willardiine derivatives: Role of the substituent at the 5-position of the uracil ring in the development of highly potent and selective GLUK5 kainate receptor antagonists. J. Med. Chem. 2007, 50, 1558–1570.
- Jane, D.E.; Lodge, D.; Collingridge, G.L. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 2009, 56, 90–113.
- Perrais, D.; Pinheiro, P.S.; Jane, D.E.; Mulle, C. Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology 2009, 56, 131–140.
- Lique Peret, A.; Christie, L.A.; Ouedraogo, D.W.; Gorlewicz, A.; Rô Me Epsztein, J.; Mulle, C.; Rie Cré Pel, V. Contribution of aberrant GluK2-containing kainate receptors to chronic seizures in temporal lobe epilepsy. Cell Rep. 2014, 8, 347–354.
- Stayte, S.; Laloli, K.J.; Rentsch, P.; Lowth, A.; Li, K.M.; Pickford, R.; Vissel, B. The kainate receptor antagonist UBP310 but not single deletion of GluK1, GluK2, or GluK3 subunits, inhibits MPTP-induced degeneration in the mouse midbrain. Exp. Neurol. 2020, 323, 113062.
- Pollok, S.; Reiner, A. Subunit-selective iGluR antagonists can potentiate heteromeric receptor responses by blocking desensitization. Proc. Natl. Acad. Sci. USA 2020, 117, 25851–25858.
- Dargan, S.L.; Clarke, V.R.J.; Alushin, G.M.; Sherwood, J.L.; Nisticò, R.; Bortolotto, Z.A.; Ogden, A.M.; Bleakman, D.; Doherty, A.J.; Lodge, D.; et al. ACET is a highly potent and specific kainate receptor antagonist: Characterisation and effects on hippocampal mossy fibre function. Neuropharmacology 2009, 56, 121–130.
- Gilron, I.; Max, M.B.; Lee, G.; Booher, S.L.; Sang, C.N.; Chappell, A.S.; Dionne, R.A. Effects of the 2-amino-3-hydroxy-5-methyl-4-isoxazole-proprionic acid/kainate antagonist LY293558 on spontaneous and evoked postoperative pain. Clin. Pharmacol. Ther. 2000, 68, 320–327.
- Sang, C.N.; Hostetter, M.P.; Gracely, R.H.; Chappell, A.S.; Schoepp, D.D.; Lee, G.; Whitcup, S.; Caruso, R.; Max, M.B. AMPA/kainate antagonist LY293558 reduces capsaicin-evoked hyperalgesia but not pain in normal skin in humans. Anesthesiology 1998, 89, 1060–1067.
- Lee, H.J.; Pogatzki-Zahn, E.M.; Brennan, T.J. The effect of the AMPA/kainate receptor antagonist LY293558 in a rat model of postoperative pain. J. Pain Palliat. Care Pharmacother. 2006, 7, 768–777.
- Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Apland, J.P.; Braga, M.F. Targeting the glutamatergic system to counteract organophosphate poisoning: A novel therapeutic strategy. Neurobiol. Dis. 2020, 133, 104406.
- Figueiredo, T.H.; Qashu, F.; Apland, J.P.; Aroniadou-Anderjaska, V.; Souza, A.P.; Braga, M.F.M. The GluK1 (GluR5) kainate/ -amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid receptor antagonist LY293558 reduces soman-induced seizures and neuropathology. J. Pharmacol. Exp. Ther. 2011, 336, 303–312.
- Martinez-Perez, J.A.; Iyengar, S.; Shannon, H.E.; Bleakman, D.; Alt, A.; Arnold, B.M.; Bell, M.G.; Bleisch, T.J.; Castaño, A.M.; Del Prado, M.; et al. GluK1 antagonists from 6-(carboxy)phenyl decahydroisoquinoline derivatives. SAR and evaluation of a prodrug strategy for oral efficacy in pain models. Bioorg. Med. Chem. Lett. 2013, 23, 6459–6462.
- O’Neill, M.J.; Bogaert, L.; Hicks, C.A.; Bond, A.; Ward, M.A.; Ebinger, G.; Ornstein, P.L.; Michotte, Y.; Lodge, D. LY377770, a novel iGlu5 kainate receptor antagonist with neuroprotective effects in global and focal cerebral ischaemia. Neuropharmacology 2000, 39, 1575–1588.
- Smolders, I.; Bortolotto, Z.A.; Clarke, V.R.; Warre, R.; Khan, G.M.; O’Neill, M.J.; Ornstein, P.L.; Bleakman, D.; Ogden, A.; Weiss, B.; et al. Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat. Neurosci. 2002, 5, 796–804.
- Simmons, R.M.; Li, D.L.; Hoo, K.H.; Deverill, M.; Ornstein, P.L.; Iyengar, S. Kainate GluR5 receptor subtype mediates the nociceptive response to formalin in the rat. Neuropharmacology 1998, 37, 25–36.
- Palecek, J.; Neugebauer, V.; Carlton, S.M.; Iyengar, S.; Willis, W.D. The effect of a kainate GluR5 receptor antagonist on responses of spinothalamic tract neurons in a model of peripheral neuropathy in primates. Pain 2004, 111, 151–161.
- Filla, S.A.; Winter, M.A.; Johnson, K.W.; Bleakman, D.; Bell, M.G.; Bleisch, T.J.; Castaño, A.M.; Clemens-Smith, A.; Del Prado, M.; Dieckman, D.K.; et al. Ethyl (3s,4aR,6S,8aR)-6-(4-ethoxycar-bonylimidazol-1-ylmethyl) decahydroiso-quinoline-3-carboxylic ester: A prodrug of a GluR5 kainate receptor antagonist active in two animal models of acute migraine. J. Med. Chem. 2002, 45, 4383–4386.
- Weiss, B.; Alt, A.; Ogden, A.M.; Gates, M.; Dieckman, D.K.; Clemens-Smith, A.; Ho, K.H.; Jarvie, K.; Rizkalla, G.; Wright, R.A.; et al. Pharmacological characterization of the competitive GLUK5 receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J. Pharmacol. Exp. Ther. 2006, 318, 772–781.
- Alushin, G.M.; Jane, D.; Mayer, M.L. Binding site and ligand flexibility revealed by high resolution crystal structures of GluK1 competitive antagonists. Neuropharmacology 2011, 60, 126–134.
- Dominguez, E.; Iyengar, S.; Shannon, H.E.; Bleakman, D.; Alt, A.; Arnold, B.M.; Bell, M.G.; Bleisch, T.J.; Buckmaster, J.L.; Castano, A.M.; et al. Two prodrugs of potent and selective GluR5 kainate receptor antagonists actives in three animal models of pain. J. Med. Chem. 2005, 48, 4200–4203.
- Martinez-Perez, J.A.; Iyengar, S.; Shannon, H.E.; Bleakman, D.; Alt, A.; Clawson, D.K.; Arnold, B.M.; Bell, M.G.; Bleisch, T.J.; Castaño, A.M.; et al. GluK1 antagonists from 6-(tetrazolyl)phenyl decahydroisoquinoline derivatives: In vitro profile and in vivo analgesic efficacy. Bioorg. Med. Chem. Lett. 2013, 23, 6463–6466.
- Chappell, A.S.; Iyengar, S.; Lobo, E.D.; Prucka, W.R. Results from clinical trials of a selective ionotropic glutamate receptor 5 (iGluR5) antagonist, LY5454694 tosylate, in 2 chronic pain conditions. Pain 2014, 155, 1140–1149.
- Madsen, U.; Bang-Andersen, B.; Brehm, L.; Christensen, I.T.; Ebert, B.; Kristoffersen, I.T.; Lang, Y.; Krogsgaard-Larsen, P. Synthesis and pharmacology of highly selective carboxy and phosphono isoxazole amino acid AMPA receptor antagonists. J. Med. Chem. 1996, 39, 1682–1691.
- Moller, E.H.; Egebjerg, J.; Brehm, L.; Stensbol, T.B.; Johansen, T.N.; Madsen, U.; Krogsgaard-Larsen, P. Resolution, absolute stereochemistry, and enantiopharmacology of the GluR1-4 and GluR5 antagonist 2-amino-3-propionic acid. Chirality 1999, 11, 752–759.
- Hald, H.; Naur, P.; Pickering, D.S.; Sprogøe, D.; Madsen, U.; Timmermann, D.B.; Ahring, P.K.; Liljefors, T.; Schousboe, A.; Egebjerg, J.; et al. Partial agonism and antagonism of the ionotropic glutamate receptor iGLuR5: Structures of the ligand-binding core in complex with domoic acid and 2-amino-3-propionic acid. J. Biol. Chem. 2007, 282, 25726–25736.
- Szymańska, E.; Frydenvang, K.; Contreras-Sanz, A.; Pickering, D.S.; Frola, E.; Serafimoska, Z.; Nielsen, B.; Kastrup, J.S.; Johansen, T.N. A new phenylalanine derivative acts as an antagonist at the AMPA receptor GluA2 and introduces partial domain closure: Synthesis, resolution, pharmacology, and crystal structure. J. Med. Chem. 2011, 54, 7289–7298.
- Venskutonyte, R.; Frydenvang, K.; Valades, E.A.; Szymańska, E.; Johansen, T.N.; Kastrup, J.S.; Pickering, D.S. Structural and pharmacological characterization of phenylalanine-based AMPA receptor antagonists at kainate receptors. ChemMedChem 2012, 7, 1793–1798.
- Szymanska, E.; Pickering, D.S.; Nielsen, B.; Johansen, T.N. 3-Substituted phenylalanines as selective AMPA- and kainate receptor ligands. Bioorg. Med. Chem. 2009, 17, 6390–6401.
- Szymańska, E.; Frydenvang, K.; Pickering, D.S.; Krintel, C.; Nielsen, B.; Kooshki, A.; Zachariassen, L.G.; Olsen, L.; Kastrup, J.S.; Johansen, T.N. Studies on aryl-substituted phenylalanines: Synthesis, activity, and different binding modes at AMPA receptors. J. Med. Chem. 2016, 59, 448–461.
- Szymańska, E.; Chałupnik, P.; Szczepańska, K.; Cuñado Moral, A.M.; Pickering, D.S.; Nielsen, B.; Johansen, T.N.; Kieć-Kononowicz, K. Design, synthesis and structure-activity relationships of novel phenylalanine-based amino acids as kainate receptors ligands. Bioorg. Med. Chem. Lett. 2016, 26, 5568–5572.
- Larsen, A.M.; Venskutonytè, R.; Valadés, E.A.; Nielsen, B.; Pickering, D.S.; Bunch, L. Discovery of a new class of ionotropic glutamate receptor antagonists by the rational design of (2 S,3 R)-3-(3-carboxyphenyl)-pyrrolidine-2-carboxylic acid. ACS Chem. Neurosci. 2011, 2, 107–114.
- Krogsgaard-Larsen, N.; Storgaard, M.; Møller, C.; Demmer, C.S.; Hansen, J.; Han, L.; Monrad, R.N.; Nielsen, B.; Tapken, D.; Pickering, D.S.; et al. Structure-activity relationship study of ionotropic glutamate receptor antagonist (2S,3R)-3-(3-carboxyphenyl)pyrrolidine-2-carboxylic acid. J. Med. Chem. 2015, 58, 6131–6150.
- Krogsgaard-Larsen, N.; Delgar, C.G.; Koch, K.; Brown, P.M.G.E.; Møller, C.; Han, L.; Huynh, T.H.V.; Hansen, S.W.; Nielsen, B.; Bowie, D.; et al. Design and synthesis of a series of l-trans-4-substituted prolines as selective antagonists for the ionotropic glutamate receptors including functional and X-ray crystallographic studies of new subtype selective kainic acid receptor subtype 1 (GluK1) antagonist (2S,4R)-4-(2-carboxyphenoxy)pyrrolidine-2-carboxylic acid. J. Med. Chem. 2017, 60, 441–457.
- Bunch, L. Pyrrolidine-2-Carboxylic Acid Derivatives as iGluR Antagonists. US20140235576A1, 21 August 2014.
- Bunch, L. Substituted 4-Proline Derivatives as iGluR Antagonists. WO2015003723A1, 15 January 2015.
- Sanders, J.M.; Ito, K.; Settimo, L.; Pentikäinen, O.T.; Shoji, M.; Sasaki, M.; Johnson, M.S.; Sakai, R.; Swanson, G.T. Divergent pharmacological activity of novel marine-derived excitatory amino acids on glutamate receptors. J. Pharmacol. Exp. Ther. 2005, 314, 1068–1078.
- Sanders, J.M.; Pentikäinen, O.T.; Settimo, L.; Pentikäinen, U.; Shoji, M.; Sasaki, M.; Sakai, R.; Johnson, M.S.; Swanson, G.T. Determination of binding site residues responsible for the subunit selectivity of novel marine-derived compounds on kainate receptors. Mol. Pharmacol. 2006, 69, 1849–1860.
- Frydenvang, K.; Lash, L.L.; Naur, P.; Postila, P.A.; Pickering, D.S.; Smith, C.M.; Gajhede, M.; Sasaki, M.; Sakai, R.; Pentikänen, O.T.; et al. Full domain closure of the ligand-binding core of the ionotropic glutamate receptor iGluR5 induced by the high affinity agonist dysiherbaine and the functional antagonist 8,9-dideoxyneodysiherbaine. J. Biol. Chem. 2009, 284, 14219–14229.
- Qiu, C.-S.; Lash-Van Wyhe, L.; Sasaki, M.; Sakai, R.; Swanson, G.T.; Gereau, R.W. Antinociceptive effects of MSVIII-19, a functional antagonist of the GluK1 kainate receptor. Pain 2011, 152, 1052–1060.
- Lash, L.L.; Sanders, J.M.; Akiyama, N.; Shoji, M.; Postila, P.; Pentikäinen, O.T.; Sasaki, M.; Sakai, R.; Swanson, G.T. Novel analogs and stereoisomers of the marine toxin neodysiherbaine with specificity for kainate receptors. J. Pharmacol. Exp. Ther. 2008, 324, 484–496.
- Swanson, G.T.; Lash, L.; Sakai, R. Kainate Receptor-Selective Epimeric Analogs of Dysiherbaine. U.S. Patent US20090118358A1, 5 July 2009.
- Brogi, S.; Brindisi, M.; Butini, S.; Kshirsagar, G.U.; Maramai, S.; Chemi, G.; Gemma, S.; Campiani, G.; Novellino, E.; Fiorenzani, P.; et al. (S)-2-Amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic acid (AMPA) and kainate receptor ligands: Further exploration of bioisosteric replacements and structural and biological investigation. J. Med. Chem. 2018, 61, 2124–2130.
- Venskutonyte, R.; Butini, S.; Coccone, S.S.; Gemma, S.; Brindisi, M.; Kumar, V.; Guarino, E.; Maramai, S.; Valenti, S.; Amir, A.; et al. Selective kainate receptor (GluK1) ligands structurally based upon 1H-cyclopentapyrimidin-2,4(1H3H)- dione: Synthesis, molecular modeling, and pharmacological and biostructural characterization. J. Med. Chem. 2011, 54, 4793–4805.
- Poulie, C.B.M.; Larsen, Y.; Leteneur, C.; Barthet, G.; Bjørn-Yoshimoto, W.E.; Malhaire, F.; Nielsen, B.; Pin, J.-P.; Mulle, C.; Pickering, D.S.; et al. (S)-2-Mercaptohistidine: A first selective orthosteric GluK3 antagonist. ACS Chem. Neurosci. 2022, 13, 1580–1587.
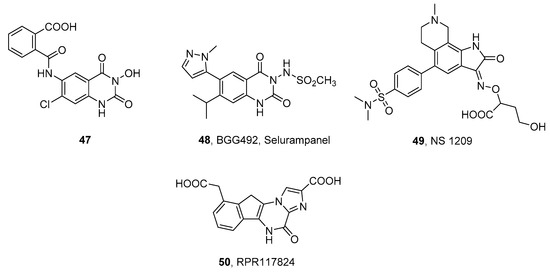
- Mula, M. Emerging drugs for focal epilepsy. Expert Opin. Emerg. Drugs 2013, 18, 87–95.
- Faught, E. BGG492 (selurampanel), an AMPA/kainate receptor antagonist drug for epilepsy. Expert Opin. Investig. Drugs 2014, 23, 107–113.
- Orain, D.; Tasdelen, E.; Haessig, S.; Koller, M.; Picard, A.; Dubois, C.; Lingenhoehl, K.; Desrayaud, S.; Floersheim, P.; Carcache, D.; et al. Design and synthesis of Selurampanel, a novel orally active and competitive AMPA receptor antagonist. ChemMedChem 2017, 12, 197–201.
- Nielsen, E.O.; Varming, T.; Mathiesen, C.; Jensen, L.H.; Moller, A.; Gouliaev, A.H.; Wätjen, F.; Drejer, J. SPD 502: A water-soluble and in vivo long-lasting AMPA antagonist with neuroprotective activity. J. Pharmacol. Exp. Ther. 1999, 289, 1492–1501.
- Keppel Hesselink, J.M. NS1209/SPD 502, a novel selective AMPA antagonist for stroke, neuropathic pain or epilepsy? Drug development lessons learned. Drug Dev. Res. 2017, 78, 75–80.
- Mignani, S.; Bohme, G.A.; Birraux, G.; Boireau, A.; Jimonet, P.; Damour, D.; Genevois-Borella, A.; Debono, M.W.; Pratt, J.; Vuilhorgne, M.; et al. 9-Carboxymethyl-5H,10H-imidazoindenopyrazin-4-one-2-carbocylic acid (RPR117824): Selective anticonvulsive and neuroprotective AMPA antagonist. Bioorg. Med. Chem. 2002, 10, 1627–1637.
- Krampfl, K.; Schlesinger, F.; Cordes, A.L.; Bufler, J. Molecular analysis of the interaction of the pyrazine derivatives RPR119990 and RPR117824 with human AMPA-type glutamate receptor channels. Neuropharmacology 2006, 50, 479–490.
- Gormsen, L.; Finnerup, N.B.; Almqvist, P.M.; Jensen, T.S. The efficacy of the AMPA receptor antagonist ns1209 and lidocaine in nerve injury pain: A randomized, double-blind, placebo-controlled, three-way crossover study. Anesth. Analg. 2009, 108, 1311–1319.
- Colotta, V.; Catarzi, D.; Varano, F.; Lenzi, O.; Filacchioni, G.; Costagli, C.; Galli, A.; Ghelardini, C.; Galeotti, N.; Gratteri, P.; et al. Structural investigation of the 7-chloro-3-hydroxy-1H-quinazoline-2,4-dione scaffold to obtain AMPA and kainate receptor selective antagonists. Synthesis, pharmacological, and molecular modeling studies. J. Med. Chem. 2006, 49, 6015–6026.
- Colotta, V.; Lenzi, O.; Catarzi, D.; Varano, F.; Squarcialupi, L.; Costagli, C.; Galli, A.; Ghelardini, C.; Pugliese, A.M.; Maraula, G.; et al. 3-Hydroxy-1H-quinazoline-2,4-dione derivatives as new antagonists at ionotropic glutamate receptors: Molecular modeling and pharmacological studies. Eur. J. Med. Chem. 2012, 54, 470–482.
- Lerma, J.; Paternain, A.V.; Rodríguez-Moreno, A.; López-García, J.C. Molecular physiology of kainate receptors. Physiol. Rev. 2001, 81, 971–998.
- Wilding, T.J.; Huettner, J.E. Activation and desensitization of hippocampal kainate receptors. J. Neurosci. 1997, 17, 2713–2721.
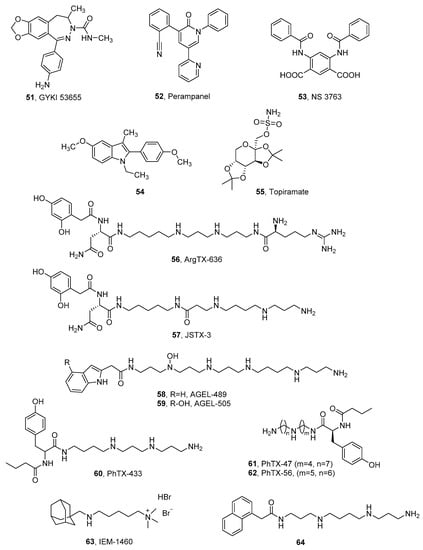
- Shvarts, V.; Chung, S. Perampanel: Newly approved, novel antiepileptic medication for partial-onset seizures. Expert Rev. Neurother. 2013, 13, 131–134.
- Fukushima, K.; Hatanaka, K.; Sagane, K.; Ido, K. Inhibitory effect of anti-seizure medications on ionotropic glutamate receptors: Special focus on AMPA receptor subunits. Epilepsy Res. 2020, 167, 106452.
- Taniguchi, S.; Stolz, J.R.; Swanson, G.T. The antiseizure drug Perampanel is a subunit-selective negative allosteric modulator of kainate receptors. J. Neurosci. 2022, 42, 5499–5509.
- Kanemura, H.; Sano, F.; Aihara, M. Usefulness of perampanel with concomitant levetiracetam for patients with drug-resistant epilepsy. Eur. J. Paediatr. Neurol. 2019, 23, 197–203.
- Trigg, A.; Brohan, E.; Cocks, K.; Jones, A.; Tahami Monfared, A.A.; Chabot, I.; Meier, G.; Campbell, R.; Li, H.; Ngo, L.Y. Health-related quality of life in pediatric patients with partial onset seizures or primary generalized tonic-clonic seizures receiving adjunctive perampanel. Epilepsy Behav. 2021, 118, 107938.
- Mehndiratta, M.M.; Manoj, G.; Jabeen, S.A.; Patten, A. Efficacy and safety of adjunctive perampanel in patients with focal seizures or generalized tonic-clonic seizures: Post hoc analysis of Phase II and Phase III double-blind and open-label extension studies in India. Epilepsia Open 2021, 8, 90–101.
- Fogarasi, A.; Flamini, R.; Milh, M.; Phillips, S.; Yoshitomi, S.; Patten, A.; Takase, T.; Laurenza, A.; Ngo, L.Y. Open-label study to investigate the safety and efficacy of adjunctive perampanel in pediatric patients. Epilepsia 2020, 61, 125–137.
- Nishida, T.; Lee, S.K.; Wu, T.; Tiamkao, S.; Dash, A. Efficacy and safety of perampanel in generalized and focal to bilateral tonic-clonic seizures: A comparative study of Asian and non-Asian populations. Epilepsia 2019, 60, 47–59.
- Renfroe, J.B.; Mintz, M.; Davis, R.; Ferreira, J.; Dispoto, S.; Ferry, J.; Umetsu, Y.; Rege, B.; Majid, O.; Hussein, Z.; et al. Adjunctive Perampanel oral suspension in pediatric patients from 2 to <12 years of age with epilepsy: Pharmacokinetics, safety, tolerability, and efficacy. J. Child Neurol. 2019, 34, 284–294.
- Usui, N.; Akamatsu, N.; Nakasato, N.; Ohnishi, A.; Kaneko, S.; Hiramatsu, H.; Saeki, K.; Miyagishi, H.; Inoue, Y. Long-term tolerability, safety and efficacy of adjunctive perampanel in the open-label, dose-ascending Study 231 and extension Study 233 in Japanese patients with epilepsy. Seizure 2018, 62, 26–32.
- Lin, K.L.; Lin, J.J.; Chou, M.L.; Hung, P.C.; Hsieh, M.Y.; Chou, I.J.; Lim, S.N.; Wu, T.; Wang, H.S. Efficacy and tolerability of perampanel in children and adolescents with pharmacoresistant epilepsy: The first real-world evaluation in Asian pediatric neurology clinics. Epilepsy Behav. 2018, 85, 188–194.
- Piña-Garza, J.E.; Lagae, L.; Villanueva, V.; Renfroe, J.B.; Laurenza, A.; Williams, B.; Kumar, D.; Meador, K.J. Long-term effects of adjunctive perampanel on cognition in adolescents with partial seizures. Epilepsy Behav. 2018, 83, 50–58.
- Eggert, K.; Squillacote, D.; Barone, P.; Dodel, R.; Katzenschlager, R.; Emre, M.; Lees, A.J.; Rascol, O.; Poewe, W.; Tolosa, E.; et al. Safety and efficacy of perampanel in advanced parkinson’s disease: A randomized, placebo-controlled study. Mov. Disord. 2010, 25, 896–905.
- Rascol, O.; Barone, P.; Behari, M.; Emre, M.; Giladi, N.; Olanow, C.W.; Ruzicka, E.; Bibbiani, F.; Squillacote, D.; Patten, A.; et al. Perampanel in Parkinson disease fluctuations: A double-blind randomized trial with placebo and entacapone. Clin. Neuropharmacol. 2012, 35, 15–20.
- Oskarsson, B.; Mauricio, E.A.; Shah, J.S.; Li, Z.; Rogawski, M.A. Cortical excitability threshold can be increased by the AMPA blocker Perampanel in amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, 215–219.
- Aizawa, H.; Kato, H.; Oba, K.; Kawahara, T.; Okubo, Y.; Saito, T.; Naito, M.; Urushitani, M.; Tamaoka, A.; Nakamagoe, K.; et al. Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J. Neurol. 2022, 269, 885–896.
- Wilding, T.J.; Chai, Y.H.; Huettner, J.E. Inhibition of rat neuronal kainate receptors by cis-unsaturated fatty acids. J. Physiol. 1998, 513 Pt 2, 331–339.
- Wilding, T.J.; Zhou, Y.; Huettner, J.E. Q/R site editing controls kainate receptor inhibition by membrane fatty acids. J. Neurosci. 2005, 25, 9470–9478.
- Wilding, T.J.; Fulling, E.; Zhou, Y.; Huettner, J.E. Amino acid substitutions in the pore helix of GluR6 control inhibition by membrane fatty acids. J. Gen. Physiol. 2008, 132, 85–99.
- Christensen, J.K.; Paternain, A.V.; Selak, S.; Ahring, P.K.; Lerma, J. A mosaic of functional kainate receptors in hippocampal interneurons. J. Neurosci. 2004, 24, 8986–8993.
- Xu, J.; Liu, Y.; Zhang, G.Y. Neuroprotection of GluR5-containing kainate receptor activation against ischemic brain injury through decreasing tyrosine phosphorylation of N-methyl-D-aspartate receptors mediated by Src kinase. J. Biol. Chem. 2008, 283, 29355–29366.
- Christensen, J.K.; Varming, T.; Ahring, P.K.; Jørgensen, T.D.; Nielsen, E. In vitro characterization of 5-carboxyl-2,4-di-benzamidobenzoic acid (NS3763), a noncompetitive antagonist of GLUK5 receptors. J. Pharmacol. Exp. Ther. 2004, 309, 1003–1010.
- Valgeirsson, J.; Nielsen, E.; Peters, D.; Varming, T.; Mathiesen, C.; Kristensen, A.S.; Madsen, U. 2-arylureidobenzoic acids: Selective noncompetitive antagonists for the homomeric kainate receptor subtype GluR5. J. Med. Chem. 2003, 46, 5834–5843.
- Valgeirsson, J.; Nielsen, E.; Peters, D.; Mathiesen, C.; Kristensen, A.S.; Madsen, U. Bioisosteric modifications of 2-arylureidobenzoic acids: Selective noncompetitive antagonists for the homomeric kainate receptor subtype GluR5. J. Med. Chem. 2004, 47, 6948–6957.
- Anna Kaczor, A.; Kronbach, C.; Unverferth, K.; Pihlaja, K.; Wiinamaki, K.; Sinkkonen, J.; Kijkowska-Murak, U.; Wrobel, T.; Stachal, T.; Matosiuk, D. Novel non-competitive antagonists of kainate GluK1/GluK2 receptors. Lett. Drug Des. Discov. 2012, 9, 891–898.
- Kaczor, A.A.; Karczmarzyk, Z.; Fruziński, A.; Pihlaja, K.; Sinkkonen, J.; Wiinämaki, K.; Kronbach, C.; Unverferth, K.; Poso, A.; Matosiuk, D. Structural studies, homology modeling and molecular docking of novel non-competitive antagonists of GluK1/GluK2 receptors. Bioorg. Med. Chem. 2014, 22, 787–795.
- Bartyzel, A.; Kaczor, A.A.; Mahmoudi, G.; Masoudiasl, A.; Wróbel, T.M.; Pitucha, M.; Matosiuk, D. Experimental and computational structural studies of 2,3,5-trisubstituted and 1,2,3,5-tetrasubstituted indoles as non-competitive antagonists of GluK1/GluK2 receptors. Molecules 2022, 27, 2479.
- Ben-Menachem, E. Topiramate: Current status and therapeutic potential. Expert Opin. Investig. Drugs 1997, 6, 1085–1094.
- Kaminski, R.M.; Banerjee, M.; Rogawski, M.A. Topiramate selectively protects against seizures induced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology 2004, 46, 1097–1104.
- Braga, M.F.M.; Aroniadou-Anderjaska, V.; Li, H.; Rogawski, M.A. Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABA_A receptors on principal neurons. J. Pharmacol. Exp. Ther. 2009, 330, 558–566.
- Gryder, D.S.; Rogawski, M.A. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J. Neurosci. 2003, 23, 7069–7074.
- Available online: www.clinicaltrials.gov (accessed on 8 December 2022).
- Strømgaard, K.; Andersen, K.; Krogsgaard-Larsen, P.; Jaroszewski, J.W. Recent advances in the medicinal chemistry of polyamine toxins. Mini-Rev. Med. Chem. 2001, 1, 317–338.
- Brown, P.M.; Aurousseau, M.R.; Musgaard, M.; Biggin, P.C.; Bowie, D. Kainate receptor pore-forming and auxiliary subunits regulate channel block by a novel mechanism. J. Physiol. 2016, 594, 1821–1840.
- Perrais, D.; Coussen, F.; Mulle, C. Atypical functional properties of GluK3-containing kainate receptors. J. Neurosci. 2009, 29, 15499–15510.
- Lomeli, H.; Wisden, W.; Köhler, M.; Keinänen, K.; Sommer, B.; Seeburg, P.H. High-affinity kainate and domoate receptors in rat brain. FEBS Lett. 1992, 307, 139–143.
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 298–487.
- Bowie, D. Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J. Biol. Chem. 2018, 293, 18789–18802.
- Mott, D.D.; Washburn, M.S.; Zhang, S.; Dingledine, R.J. Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J. Neurosci. 2003, 23, 1179–1188.
- Quistad, G.B.; Suwanrumpha, S.; Jarema, M.A.; Shapiro, M.J.; Skinner, W.S.; Jamieson, G.C.; Lui, A.; Fu, E.W. Structure of paralytic acylpolyamines from the spider Agelenopsisaperta. Biochem. Biophys. Res. Commun. 1990, 169, 51–56.
- Eldefrawi, A.T.; Eldefrawi, M.E.; Konnot, K.; Mansour, N.A.; Nakanishit, K.; Oltzt, E.; Usherwood, P.N.R. Structure and synthesis of a potent glutamate receptor antagonist in wasp venom (wasp venom toxin/quisqualate receptor). Proc. Natl. Acad. Sci. USA 1988, 85, 4910–4913.
- Kachel, H.S.; Franzyk, H.; Mellor, I.R. Philanthotoxin analogues that selectively inhibit ganglionic nicotinic acetylcholine receptors with exceptional potency. J. Med. Chem. 2019, 62, 6214–6222.
- Vassileiou, C.; Kalantzi, S.; Vachlioti, E.; Athanassopoulos, C.M.; Koutsakis, C.; Piperigkou, Z.; Karamanos, N.; Stivarou, T.; Lymberi, P.; Avgoustakis, K.; et al. New analogs of polyamine toxins from spiders and wasps: Liquid phase fragment synthesis and evaluation of antiproliferative activity. Molecules 2022, 27, 447.
- Verdoni, M.; Roudaut, H.; De Pomyers, H.; Gigmes, D.; Bertin, D.; Luis, J.; Bengeloune, A.H.; Mabrouk, K. ArgTX-636, a polyamine isolated from spider venom: A novel class of melanogenesis inhibitors. Bioorg. Med. Chem. 2016, 24, 5685–5692.
- Kromann, H.; Krikstolaityte, S.; Andersen, A.J.; Andersen, K.; Krogsgaard-Larsen, P.; Jaroszewski, J.W.; Egebjerg, J.; Strømgaard, K. Solid-phase synthesis of polyamine toxin analogues: Potent and selective antagonists of Ca2+-permeable AMPA receptors. J. Med. Chem. 2002, 45, 5745–5754.
- Kachel, H.S.; Patel, R.N.; Franzyk, H.; Mellor, I.R. Block of nicotinic acetylcholine receptors by philanthotoxins is strongly dependent on their subunit composition. Sci. Rep. 2016, 6, 38116.
- Matavel, A.; Estrada, G.; De Marco Almeida, F. Spider venom and drug discovery: A review. In Spider Venoms; Springer: Dordrecht, The Netherlands, 2016; pp. 273–292.
- Magazanik, L.G.; Buldakova, S.L.; Samoilova, M.V.; Gmiro, V.E.; Mellor, I.R.; Usherwood, P.N.R. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J. Physiol. 1997, 505 Pt 3, 655–663.
- Koike, M.; Iino, M.; Ozawa, S. Blocking effect of 1-naphthyl acetyl spermine on Ca2+-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci. Res. 1997, 29, 27–36.
- Nelson, J.K.; Frølund, S.U.; Tikhonov, D.B.; Kristensen, A.S.; Strømgaard, K. Synthesis and biological activity of argiotoxin 636 and analogues: Selective antagonists for ionotropic glutamate receptors. Angew. Chem. Int. Ed. 2009, 48, 3087–3091.
- Xiong, X.F.; Poulsen, M.H.; Hussein, R.A.; Nørager, N.G.; Strømgaard, K. Structure-activity relationship study of spider polyamine toxins as inhibitors of ionotropic glutamate receptors. ChemMedChem 2014, 9, 2661–2670.
- Nørager, N.G.; Poulsen, M.H.; Jensen, A.G.; Jeppesen, N.S.; Kristensen, A.S.; Strømgaard, K. Structure−activity relationship studies of n-methylated and n-hydroxylated spider polyamine toxins as inhibitors of ionotropic glutamate receptors. J. Med. Chem. 2014, 57, 4940–4949.
- Poulsen, M.H.; Lucas, S.; Bach, T.B.; Barslund, A.F.; Wenzler, C.; Jensen, C.B.; Kristensen, A.S.; Strømgaard, K. Structure−Activity relationship studies of argiotoxins: Selective and potent inhibitors of ionotropic glutamate receptors. J. Med. Chem. 2013, 56, 1171–1181.