Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ivano Mezzaroma and Version 2 by Camila Xu.
Systemic chronic immune activation and CD4+ T-cell depletion characterize the progression of human immunodeficiency virus type-1 (HIV-1) infection toward acquired immune deficiency syndrome (AIDS).
- HIV-1
- AIDS
- inflammaging
- immune activation
1. Introduction
Systemic chronic immune activation and CD4+ T-cell depletion characterize the progression of human immunodeficiency virus type-1 (HIV-1) infection toward acquired immune deficiency syndrome (AIDS). However, the causal link between these two phenomena has not been formally established. Persistent activation is observed in numerous components of both the innate and adaptative immune system, including cells (e.g., activated phenotypes of macrophages and dendritic cells), cytokines and chemokines [tumor necrosis factor, interleukin (IL)-1, IL-6, IL-8, IL-15, and IL-10], acute phase proteins (serum amyloid A, C-reactive protein), elements of the coagulation cascade (D-dimers, tissue factor), elements of fibrosis (matrix metalloproteinase activation, collagen deposition), and microbial sensors (lipopolysaccharide binding protein, soluble CD14). Increased turnover and exhaustion of T cells and turnover of B cells are observed with an altered phenotypic profile and hypergammaglobulinemia [1]. Moreover, high levels of systemic immune activation and inflammation not only promote viral replication and CD4+ T-cell apoptosis, but they may also lead to a more rapid decline of immune function and competence, which have been associated with aging [2].
With the introduction of combined antiretroviral therapy (cART), the immune responses, morbidity, and mortality of people living with HIV (PLWH) have significantly improved. The life expectancy of PLWH has dramatically increased and is only slightly shorter than that of uninfected individuals, so the effects of aging on HIV-1-positive patients have begun to be evident [3]. Several disorders that typically affect the aged population now appear in relatively young HIV-1 subjects, such as neurocognitive disorders, cardiovascular diseases (CVD), metabolic syndrome (MS), bone abnormalities, and non-HIV-1-associated cancers [4][5]. The Strategies for Management of Antiretroviral Therapy (SMART) study demonstrated that non-AIDS-defining age-related comorbidities are the major cause of morbidity and mortality, compared to opportunistic diseases [6]. Most of these pathologies have been linked to immune-senescence and inflammageing, a type of premature aging present in PLWH.
 The most obvious cause of immune activation is direct antigenic stimulation by the virus and its products, such as Nef and gp120, which stimulate the activation of lymphocytes and macrophages, resulting in the secretion of pro-inflammatory cytokines and chemokines. The viral protein Nef, for example, has been shown to reduce endothelial nitric oxide production, promote secretion of endothelial-cell derived MCP-1, induce endothelial cell apoptosis, and increase inflammatory cytokine release from macrophages [29][30]. In addition, HIV-1 components also bind to pattern recognition receptors, such as Toll-like receptors (TLR) 7 and 9 [31][32]. Del Cornò and colleagues provided evidence of an interplay between HIV-1 gp120 and host TLR4 in human monocyte-derived macrophages and hepatic stellate cells, which led to intracellular pathways and biologic activities that mediate proinflammatory and profibrogenic signals. In particular, this interaction resulted in the activation of the NF-kB and MAPK pathways, leading to downstream up-regulation of pro-inflammatory cytokines and chemokines in human monocyte-derived macrophages and to cell migration and secretion of CCL2 and CXCL8 in hematopoietic stem cells [33].
The level of viremia also correlates with the level of immune activation, as shown in cART-treated subjects or ECs. It has been estimated that only approximately 20% of circulating CD8+ T cells are HIV-1 specific in untreated chronically infected patients [34], whereas HIV-1-specific CD4+ T cells are usually present at a lower magnitude, up to approximately 3% of circulating CD4+ T cells [35]. However, high levels of HIV-1 replication are insufficient to induce pathological levels of immune activation. Furthermore, recent data report that inflammation is not directly associated with the size of the blood reservoir, neither with total HIV DNA [36] nor with intact proviral DNA levels [37][38][39]. Interestingly, a strong positive correlation between HIV DNA and CD8+ T-cell activation was found in viremic patients with primary or chronic infection, whereas there was no correlation was found between T-cell activation and HIV DNA in patients with successfully treated chronic infection [40].
Latent viruses, such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV), reactivate more frequently during HIV-1 infection due to the depletion of CD4+ T cells and loss of CD8+ T cells that control viral replication. This reactivation contributes to the continued stimulation of the immune system [41][42][43]. Moreover, other viruses can also contribute to immune activation, such as hepatitis B or C viruses (HBV or HCV). In fact, different levels of inflammation exist between patients co-infected with HIV-1 and HBV or HCV compared with mono-infected and uninfected controls. Several studies have shown that HIV/HCV and HIV/HBV co-infected patients have higher levels of plasma inflammation and microbial translocation biomarkers than HIV-1 mono-infected patients and these markers were positively correlated with indices of hepatic damage [44][45][46]. In addition to such chronic viral infections, different pathogens can cause systemic immune activation and dysregulation of the immune system to a similar extent as HIV-1 infection. For example, non-HIV-associated immune activation observed in chronic helminthic infections could contribute to CD4+ T-cell loss and dysregulated immune response in PLWH co-infected with helminths, even if a specific role in disease progression has not been sufficiently demonstrated [47].
The massive depletion of CD4+ T cells in the gastrointestinal tract (GUT) during primary infection and the concomitant accumulation of inflammatory cells, such as dendritic cells, neutrophils, and monocytes, progressively compromise the mucosal integrity and disrupt the mucosal barrier [48][49]. This dysregulation favors microbial translocation, resulting in translocation of peptidoglycan, lipoteichoic acid, LPS, flagellin, and CpG DNA from the intestinal lumen into the systemic circulation. Stimulating several TLRs, the microbial products active a signaling cascade and induce the production of pro-inflammatory cytokines, such as IL-1β, IL-6, TNF-α, and type I interferons [50]. This local and systemic inflammation caused by microbial translocation contributes to aberrant immune activation in chronic HIV-1 infection.
Moreover, several studies reported that sCD14 plasma levels in PLWH were significantly higher than in HIV-1-negative subjects, but were similar among PLWH stratified according to plasma viral load, even in those with residual viral replication [51][52][53].
The most obvious cause of immune activation is direct antigenic stimulation by the virus and its products, such as Nef and gp120, which stimulate the activation of lymphocytes and macrophages, resulting in the secretion of pro-inflammatory cytokines and chemokines. The viral protein Nef, for example, has been shown to reduce endothelial nitric oxide production, promote secretion of endothelial-cell derived MCP-1, induce endothelial cell apoptosis, and increase inflammatory cytokine release from macrophages [29][30]. In addition, HIV-1 components also bind to pattern recognition receptors, such as Toll-like receptors (TLR) 7 and 9 [31][32]. Del Cornò and colleagues provided evidence of an interplay between HIV-1 gp120 and host TLR4 in human monocyte-derived macrophages and hepatic stellate cells, which led to intracellular pathways and biologic activities that mediate proinflammatory and profibrogenic signals. In particular, this interaction resulted in the activation of the NF-kB and MAPK pathways, leading to downstream up-regulation of pro-inflammatory cytokines and chemokines in human monocyte-derived macrophages and to cell migration and secretion of CCL2 and CXCL8 in hematopoietic stem cells [33].
The level of viremia also correlates with the level of immune activation, as shown in cART-treated subjects or ECs. It has been estimated that only approximately 20% of circulating CD8+ T cells are HIV-1 specific in untreated chronically infected patients [34], whereas HIV-1-specific CD4+ T cells are usually present at a lower magnitude, up to approximately 3% of circulating CD4+ T cells [35]. However, high levels of HIV-1 replication are insufficient to induce pathological levels of immune activation. Furthermore, recent data report that inflammation is not directly associated with the size of the blood reservoir, neither with total HIV DNA [36] nor with intact proviral DNA levels [37][38][39]. Interestingly, a strong positive correlation between HIV DNA and CD8+ T-cell activation was found in viremic patients with primary or chronic infection, whereas there was no correlation was found between T-cell activation and HIV DNA in patients with successfully treated chronic infection [40].
Latent viruses, such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV), reactivate more frequently during HIV-1 infection due to the depletion of CD4+ T cells and loss of CD8+ T cells that control viral replication. This reactivation contributes to the continued stimulation of the immune system [41][42][43]. Moreover, other viruses can also contribute to immune activation, such as hepatitis B or C viruses (HBV or HCV). In fact, different levels of inflammation exist between patients co-infected with HIV-1 and HBV or HCV compared with mono-infected and uninfected controls. Several studies have shown that HIV/HCV and HIV/HBV co-infected patients have higher levels of plasma inflammation and microbial translocation biomarkers than HIV-1 mono-infected patients and these markers were positively correlated with indices of hepatic damage [44][45][46]. In addition to such chronic viral infections, different pathogens can cause systemic immune activation and dysregulation of the immune system to a similar extent as HIV-1 infection. For example, non-HIV-associated immune activation observed in chronic helminthic infections could contribute to CD4+ T-cell loss and dysregulated immune response in PLWH co-infected with helminths, even if a specific role in disease progression has not been sufficiently demonstrated [47].
The massive depletion of CD4+ T cells in the gastrointestinal tract (GUT) during primary infection and the concomitant accumulation of inflammatory cells, such as dendritic cells, neutrophils, and monocytes, progressively compromise the mucosal integrity and disrupt the mucosal barrier [48][49]. This dysregulation favors microbial translocation, resulting in translocation of peptidoglycan, lipoteichoic acid, LPS, flagellin, and CpG DNA from the intestinal lumen into the systemic circulation. Stimulating several TLRs, the microbial products active a signaling cascade and induce the production of pro-inflammatory cytokines, such as IL-1β, IL-6, TNF-α, and type I interferons [50]. This local and systemic inflammation caused by microbial translocation contributes to aberrant immune activation in chronic HIV-1 infection.
Moreover, several studies reported that sCD14 plasma levels in PLWH were significantly higher than in HIV-1-negative subjects, but were similar among PLWH stratified according to plasma viral load, even in those with residual viral replication [51][52][53].
 During the chronic phase of HIV-1 infection, both the accelerated process of immune senescence and inflammaging may contribute to the development of the progressive immunodeficiency [66].
During the chronic phase of HIV-1 infection, both the accelerated process of immune senescence and inflammaging may contribute to the development of the progressive immunodeficiency [66].
2. Role of Immune Activation in Progression to AIDS
Studies of pathogenic and nonpathogenic models of simian immunodeficiency virus (SIV) infection have provided insight into the role of systemic immune activation in the progression to AIDS. Natural SIV hosts, such as African green monkeys (AGMs) and sooty mangabeys (SMs), generally are able to live normally with the virus and do not progress to immunodeficiency, despite sustained high levels of plasma viremia. On the contrary, in other non-human primates, such as rhesus macaques (RMs) and Asian pigtailed macaques, SIV infection results in immunodeficiency and progression to AIDS, similar to HIV-1 infection [7]. During both pathogenic and non-pathogenic infection, robust viral replication and early antiviral responses occur during the acute phase of infection, but they show interesting immunological differences: SMs do not exhibit the increased CD4+ T-cell turnover and generalized immune activation that is characteristic of SIV infection in RMs or HIV-1 infection in humans [8][9][10]. Natural hosts have evolved strategies to avoid disease progression and achieve an effective response, which enables symbiotic coexistence. The observed adaptations include early resolution of acute T-cell activation, rather than improved viral control. Natural hosts have the ability to attenuate acute innate immune responses to SIV after a few weeks of infection, contrary to pathogenic SIV and HIV-1 infections in which immune activation persists throughout the course of the disease. SIV-infected SMs have low levels of immune activation, T-cell turnover, and cell cycle perturbation compared to SIV-infected RMs or HIV-1-infected humans, with more comparable levels to uninfected animals [11][12][13][14]. Moreover, non-pathogenic infections do not reveal microbial translocation, as shown by lack of LPS or sCD14 in the plasma of SIV- infected RMs or AGMs [15][16], and experimentally induced immune activation with LPS in natural hosts shows significantly increased virus replication and CD4+ T-cell depletion [17]. These observations about the lack of chronic immune activation and microbial translocation in disease progression observed in natural hosts have brought attention to the role of immune activation in HIV-1 infection. The precise mechanisms underlying the resolution of acute immune activation in SIV-infected SMs and AGMs remain poorly understood and are likely quite complex. Another observation supporting the role of immune activation as the major driving force of CD4+ T-cell loss and AIDS is provided by the elite controller group (ECs). ECs represent a small subset of PLWH (about 3/1000) [18][19][20] who are able to maintain a stable CD4+ T-cell count (irrespective of a threshold) and have a viral load persistently below 50 copies/mL for more than 12 months [21], even in the absence of cART. It appears that viral genetic defects or humoral responses in ECs do not play a major role in controlling HIV-1 viral load, whereas innate responses of potential interest have been reported [22]. Interestingly, AIDS events have been described in a few ECs with loss of CD4+ T cells, despite maintaining undetectable viral loads [23][24][25]. The study of progression to AIDS in ECs has provided invaluable insight for deeper comprehension of immune mechanisms controlling HIV-1 infection and disease progression. Indeed ECs, despite spontaneously viral replication control, show higher levels of T-cell activation than healthy donors and even higher levels than cART-suppressed individuals [26]. These individuals with higher T-cell activation display slow but progressive CD4+ T-cell loss and can develop AIDS, emphasizing the role of immune activation in the pathogenesis of HIV-1 infection.3. Proposed Mechanisms Inducing Chronic Immune Activation
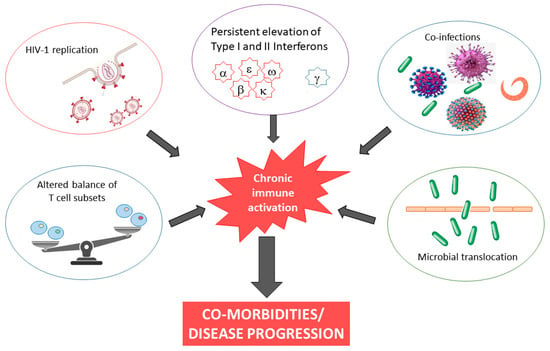
The causes of chronic inflammation and immune activation in HIV-1 infection are incompletely understood but are likely multifactorial in nature, involving both direct and indirect stimuli. Although ouresearchers' understanding remains incomplete, possible explanations include microbial translocation, co-infection, and continued presence of HIV RNA, almost always present at levels below the detection limits of clinical assays, in cART-treated subjects (Figure 1). Finally, HIV-1 infection is characterized by persistent elevation of type I and II interferons. It has been demonstrated that inadequate regulation of IFN responses drives chronic immune activation [27][28].Figure 1.
Causes and consequences of chronic immune activation in HIV-1 infection.
4. The Detrimental Consequences of Systemic Immune Activation
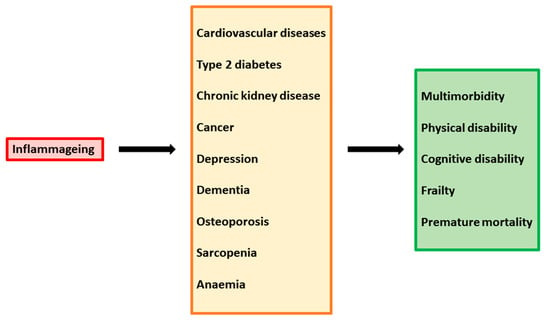
The persistent state of immune activation and inflammation in HIV-1 infection has extensive and detrimental effects on the host immune system and patient outcome. Immune system dysregulation, characterized by a shift in leukocyte activity and an imbalance in cytokine levels, plays a pathogenic role in the setting of HIV-1 infection. Since the virus preferentially infects and kills activated CD4+ T-helper cells [54], the repertoire of these cells is altered and the loss of T-cell homeostasis compromises the host’s ability to control a wide range of potential pathogens. The contemporaneous presence of high levels of CD4+ T-cell activation and high levels of viremia leads to further new infection and consequent death of CD4+ cells. Moreover, the depletion of CD4+ T cells triggers a homeostatic response of the immune system, which stimulates the activation and proliferation of surviving cells to replenish the compartment, providing further targets for the virus. This dysregulation is exacerbated by the inhibition of normal function in B-cells, NKs, and other antigen-presenting cells, as well as by the achievement of persistent replicative senescence in T cells [55][56]. In addition, elevated levels of HIV-1-associated immune activation products, such as pro-inflammatory and pro-apoptosis cytokines, sustain generalized damage to the host immune system. Indeed, many studies report uncommon levels of many cytokines, such as pro-inflammatory IL-1β, IL-2, IL-6, IL-8, and TNF-α, but also anti-inflammatory cytokines, such as IL-4, IL-10, and IL-13. Moreover, increased levels of MIP-1α, ICAM, VCAM, MCP-1, and CXCL9 were found [57][58][59][60][61][62]. Immune system dysregulation also results in inflammatory damage to the architecture of tissue involved in T-cell regeneration and function, such as bone marrow, the thymus, and lymph nodes. In particularly, altered thymic function results in suboptimal production of naïve T cells, greater differentiation of naïve cells into effector/memory cells, and hindered immune reconstitution [63][64][65]. Importantly, the chronic stimulation of the immune system and high levels of pro-inflammatory cytokines increase susceptibility to premature aging of the immune system. The phenomenon of “inflammageing” has begun to be evident in the last decades, as a consequence of increased life expectancy due to the introduction of cART. In cART-treated patients, quality of life and survival have improved substantially; however, these individuals are predisposed to chronic inflammatory conditions leading age-associated diseases, such as inflammatory bowel disease, neurocognitive disorders, cardiovascular diseases, metabolic syndrome, bone abnormalities, and non-HIV-associated cancers (Figure 2).Figure 2.
Inflammaging as a driver of multiple chronic diseases.
5. Effect of cART on HIV-1 Associated Immune Activation
Although the introduction of cART has made it possible to achieve durable control of viral replication, cART is not curative and cannot eradicate HIV-1 from the body. Antiretroviral drugs prevent the capacity of HIV-1 to replicate, which can be defined as the spread of infectious virus from one cell to another cell. These drugs do not target integrated HIV DNA and are unable to eliminate long-lived cells that harbor proviruses; thus, despite cART being fully suppressive, the virus will persist for decades. During cART, viral replication is effectively controlled in many PLWH and HIV-1 viral loads are suppressed to below detectable levels. However, if treatment is stopped, HIV-1 usually rebounds to high levels [67]. Even when cART-treated subjects have undetectable viral loads based on current clinical assays, ultrasensitive methods can still reveal HIV RNA in plasma [68]. The source of this persistent, low-level viremia remains unclear; it is probably derived from ongoing rounds of viral replication, or activation of infected resting T cells in the latent reservoir, or some combination of the two [69]. Although it is theoretically possible that residual viremia stimulates the immune system and contributes to CVD, these relationships have yet to be proven [70]. Iannetta and colleagues reported that cART was able to reduce myeloid and lymphoid inflammation in advanced and non-advanced PLWH by increasing circulating plasmacytoid cell counts and normalizing HLA-DR expression on myeloid dendritic cells and non-classical monocytes, even during the first year of treatment [71]. In cART-treated PLWH with undetected viremia, the level of inflammation markers is dramatically reduced compared to baseline; however, cART has not been successful in normalizing elevated markers of systemic immune activation [72]. For example, levels of several pro-inflammatory molecules, such as CRP, IL-6, and D-dimer [73], as well as markers of T-cell activation [74][75], remain higher in PLWH than in uninfected controls despite suppressive cART, and this increase has been associated with higher mortality [76][77][78][79][80]. Early initiation of cART, within the first 6 months of infection, seems to achieve lower levels of immune activation than when treatment is started even after a few years [81]. However, initiation of cART during acute HIV-1 infection is insufficient to resolve the chronic inflammation, and the inflammation in these patients remains higher than in uninfected controls even when cART is started early. In cART-treated patients, levels of pro-inflammatory cytokines are also associated with increased risk of CVD, independently from other CVD risk factors [82][83], and with infection-related and unrelated cancers, even after adjusting for demographics and CD4+ T cell counts [84]. In addition, higher levels of TNF-α were also found to be significantly associated with the occurrence of serious non-AIDS events [85].6. Gender Differences in HIV-1 Associated Immune Activation
In recent years, some studies have shown that sex is implicated in HIV-1 pathogenesis. Biological sex is an important contributor to disease pathogenesis in multiple infectious diseases [86], with a distinct genetic complement, hormonal environment, and behavioral and social context. Sex differences have been described for diverse aspects of HIV-1 infection and disease, including transmission, pathogenesis, morbidity, mortality, and response to antiretroviral treatment. In addition, sex difference seems to influence immune activation and HIV-1-associated co-morbidities [87]. Furthermore, sex-specific differences in CD4+ T cell counts have been reported in several studies, both in naïve and in cART-treated patients: females had higher CD4 cell counts and fewer AIDS-defining illnesses [88][89][90]. Moreover, it has been demonstrated that chronically HIV-1-infected women have significantly higher levels of CD8+ T-cell activation than men with the same HIV-1 viral load, thereby experiencing a significantly increased risk of developing AIDS compared to men with similar levels of HIV-1 replication [91]. Systemic immune activation markers have been shown to be higher in HIV-1 infected women than in HIV-1 infected men or uninfected women [92]. Higher expression of IFN-stimulated genes was observed in women. In the short term, a hyper-acute innate immune response to infection could allow women to better control viral replication; however, with time, this hyper-stimulation of the immune system could lead to a dysfunctional response. To date, the precise mechanisms responsible for these reported sex differences in viral load, T cell count, and immunological differences remain unknown. This elevated immune-activation in women could result from sex-specific environmental risk factors, sex differences in the microbiome [93], steroid hormones secreted by gonads [94], and direct effects of X and Y chromosome-linked factors [95]. Several genes on the X chromosome can potentially influence immunocompetence; in particular, the X chromosome encodes for FOXP3, the lineage-defining transcription factor of regulatory T cells; IL-2Rγ, a common cytokine receptor; and pattern recognition receptors (PRR) TLR7 and TLR8, which are known to sense HIV-1 ssRNA. Notably, X chromosome inactivation is a random process and an estimated 20% of the X chromosome escapes inactivation that, consequently, may lead to overexpression of certain gene products [96]. In addition, growing evidence supports a potential epigenetic regulation of sex differences in immune responses [97]. Other studies suggest that sex hormones influence HIV-1 acquisition through changes in microbiome composition, for example, reducing bacterial vaginosis could modulate the gut microbiome and thus contribute to systemic inflammation [98]. Sex hormones are also involved in IFN-α production since estrogen and progesterone have been reported to modulate plasmacytoid dendritic cell secretion [99][100].References
- Douek, D.C. Immune activation, HIV persistence, and the cure. Top. Antivir. Med. 2013, 21, 128–132.
- Sokoya, T.; Steel, H.C.; Nieuwoudt, M.; Rossouw, T.M. HIV as a Cause of Immune Activation and Immunosenescence. Mediat. Inflamm. 2017, 2017, 6825493.
- Nakagawa, F.; May, M.; Phillips, A. Life expectancy living with HIV: Recent estimates and future implications. Curr. Opin. Infect. Dis. 2013, 26, 17–25.
- Nasi, M.; Pinti, M.; Mussini, C.; Cossarizza, A. Persistent inflammation in HIV infection: Established concepts, new perspectives. Immunol. Lett. 2014, 161, 184–188.
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2017, 187, 44–52.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group. CD4+ count-guided interruption of antiretroviral treatment. N. Engl. J. Med. 2006, 355, 2283–2296.
- Klatt, N.R.; Silvestri, G.; Hirsch, V. Nonpathogenic Simian Immunodeficiency Virus Infections. Cold Spring Harb. Perspect. Med. 2012, 2, a007153.
- Dunham, R.; Pagliardini, P.; Gordon, S.; Sumpter, B.; Engram, J.; Moanna, A.; Paiardini, M.; Mandl, J.N.; Lawson, B.; Garg, S.; et al. The AIDS resistance of naturally SIV-infected sooty mangabeys is independent of cellular immunity to the virus. Blood 2006, 108, 209–217.
- Silvestri, G. Naturally SIV-infected sooty mangabeys: Are we closer to understanding why they do not develop AIDS? J. Med. Primatol. 2005, 34, 243–252.
- Silvestri, G.; Paiardini, M.; Pandrea, I.; Lederman, M.M.; Sodora, D.L. Understanding the benign nature of SIV infection in natural hosts. J. Clin. Investig. 2007, 117, 3148–3154.
- Silvestri, G.; Sodora, D.L.; Koup, R.A.; Paiardini, M.; O’Neil, S.P.; McClure, H.M.; Staprans, S.I.; Feinberg, M.B. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003, 18, 441–452.
- Paiardini, M.; Cervasi, B.; Sumpter, B.; McClure, H.M.; Sodora, D.L.; Magnani, M.; Staprans, S.I.; Piedimonte, G.; Silvestri, G. Perturbations of cell cycle control in T cells contribute to the different outcomes of simian immunodeficiency virus infection in rhesus macaques and sooty mangabeys. J. Virol. 2006, 80, 634–642.
- Sumpter, B.; Dunham, R.; Gordon, S.; Engram, J.; Hennessy, M.; Kinter, A.; Paiardini, M.; Cervasi, B.; Klatt, N.; McClure, H.; et al. Correlates of preserved CD4+ T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: Implications for AIDS pathogenesis. J. Immunol. 2007, 178, 1680–1691.
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371.
- Pandrea, I.V.; Gautam, R.; Ribeiro, R.M.; Brenchley, J.M.; Butler, I.F.; Pattison, M.; Rasmussen, T.; Marx, P.A.; Silvestri, G.; Lackner, A.A.; et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J. Immunol. 2007, 179, 3035–3046.
- Pandrea, I.; Gaufin, T.; Brenchley, J.M.; Gautam, R.; Monjure, C.; Gautam, A.; Coleman, C.; Lackner, A.A.; Ribeiro, R.M.; Douek, D.C.; et al. Cutting edge: Experimentally induced immune activation in natural hosts of SIV induces significant increases in viral replication and CD4+ T cell depletion. J. Immunol. 2008, 181, 6687–6691.
- Deeks, S.G.; Walker, B.D. Human immunodeficiency virus controllers: Mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 2007, 27, 406–416.
- Krishnan, S.; Wilson, E.M.P.; Sheikh, V.; Rupert, A.; Mendoza, D.; Yang, J.; Lempicki, R.; Migueles, S.A.; Sereti, I. Evidence for Innate Immune System Activation in HIV Type 1–Infected Elite Controllers. J. Infect. Dis. 2014, 209, 931–939.
- Okulicz, J.F.; Lambotte, O. Epidemiology and clinical characteristics of elite controllers. Curr. Opin. HIV AIDS 2011, 6, 163–168.
- Gonzalo-Gil, E.; Ikediobi, U.; Sutton, R.E. Mechanisms of Virologic Control and Clinical Characteristics of HIV+ Elite/Viremic Controllers. Yale J. Biol. Med. 2017, 90, 245–259.
- Genovese, L.; Nebuloni, M.; Alfano, M. Cell-Mediated Immunity in Elite Controllers Naturally Controlling HIV Viral Load. Front. Immunol. 2013, 4, 86.
- Pereyra, F.; Addo, M.M.; Kaufmann, D.E.; Liu, Y.; Miura, T.; Rathod, A.; Baker, B.; Trocha, A.; Rosenberg, R.; Mackey, E.; et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 2008, 197, 563–571.
- Pereyra, F.; Palmer, S.; Miura, T.; Block, B.L.; Wiegand, A.; Rothchild, A.C.; Baker, B.; Rosenberg, R.; Cutrell, E.; Seamanet, M.S.; et al. Persistent low-level viremia in HIV-1 elite controllers and relationship to immunologic parameters. J. Infect. Dis. 2009, 200, 984–990.
- Sajadi, M.M.; Constantine, N.T.; Mann, D.L.; Charurat, M.; Dadzan, E.; Kadlecik, P.; Redfield, R.R. Epidemiologic characteristics and natural history of HIV-1 natural viral suppressors. J. Acquir. Immune Defic. Syndr. 2009, 50, 403–408.
- Hunt, P.W.; Brenchley, J.; Sinclair, E.; McCune, J.M.; Roland, M.; Page-Shafer, K.; Hsue, P.; Emu, B.; Krone, M.; Lampiris, H.; et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J. Infect. Dis. 2008, 197, 126–133.
- Sousa, A.E.; Carneiro, J.; Meier-Schellersheim, M.; Grossman, Z.; Victorino, R.M. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 2002, 169, 3400–3406.
- Rajasuriar, R.; Khoury, G.; Kamarulzaman, A.; French, M.A.; Cameron, P.U.; Lewin, S.R. Persistent immune activation in chronic HIV infection: Do any interventions work? AIDS 2013, 27, 1199–1208.
- Duffy, P.; Wang, X.; Lin, P.H.; Yao, Q.; Chen, C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J. Surg. Res. 2009, 156, 257–264.
- Wang, T.; Green, L.A.; Gupta, S.K.; Kim, C.; Wang, L.; Almodovar, S.; Flores, S.C.; Prudovsky, I.A.; Jolicoeur, P.; Liu, Z.; et al. Transfer of Intracellular HIV Nef to Endothelium Causes Endothelial Dysfunction. PLoS ONE 2014, 9, e91063.
- Betts, M.R.; Ambrozak, D.R.; Douek, D.C.; Bonhoeffer, S.; Brenchley, J.M.; Casazza, J.P.; Koup, R.A.; Picker, L.J. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: Relationship to viral load in untreated HIV infection. J. Virol. 2001, 75, 11983–11991.
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191.
- Appay, V.; Fastenackels, S.; Katlama, C.; Ait-Mohand, H.; Schneider, L.; Guihot, A.; Keller, M.; Grubeck-Loebenstein, B.; Simon, A.; Lambotte, O.; et al. Old age and anti-cytomegalovirus immunity are associated with altered T-cell reconstitution in HIV-1-infected patients. AIDS 2011, 25, 1813–1822.
- Del Cornò, M.; Cappon, A.; Donninelli, G.; Varano, B.; Marra, F.; Gessani, S. HIV-1 gp120 signaling through TLR4 modulates innate immune activation in human macrophages and the biology of hepatic stellate cells. J. Leukoc. Biol. 2016, 100, 599–606.
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98.
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV- 1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Invest. 2005, 115, 3265–3275.
- Falasca, F.; Di Carlo, D.; De Vito, C.; Bon, I.; d’Ettorre, G.; Fantauzzi, A.; Mezzaroma, I.; Fimiani, C.; Re, M.C.; Vullo, V.; et al. Evaluation of HIV-DNA and inflammatory markers in HIV-infected individuals with different viral load patterns. BMC Infect. Dis. 2017, 17, 581.
- Olson, A.; Coote, C.; Snyder-Cappione, J.E.; Lin, N.; Sagar, M. HIV-1 Transcription but Not Intact Provirus Levels are Associated With Systemic Inflammation. J. Infect. Dis. 2021, 223, 1934–1942.
- Mexas, A.M.; Graf, E.H.; Pace, M.J.; Yu, J.J.; Papasavvas, E.; Azzoni, L.; Busch, M.P.; Di Mascio, M.; Foulkes, A.S.; Migueles, S.A.; et al. Concurrent measures of total and integrated HIV DNA monitor reservoirs and ongoing replication in eradication trials. AIDS 2012, 26, 2295–2306.
- Allavena, C.; Rodallec, A.; Sécher, S.; Reliquet, V.; Baffoin, S.; André-Garnier, E.; Billaud, E.; Raffi, F.; Ferré, V. Evaluation of residual viremia and quantitation of soluble CD14 in a large cohort of HIV-infected adults on a long-term non-nucleoside reverse transcriptase inhibitor-based regimen. J. Med. Virol. 2013, 85, 1878–1882.
- Wiesmann, F.; Braun, P.; Knickmann, M.; Knechten, H. Low level HIV viremia is more frequent under protease-inhibitor containing first line therapy than under NNRTI-regimens. J. Int. AIDS Soc. 2014, 17, 19828.
- Righetti, E.; Ballon, G.; Ometto, L.; Cattelan, A.M.; Menin, C.; Zanchetta, M.; Chieco-Bianchi, L.; De Rossi, A. Dynamics of Epstein-Barr virus in HIV-1- infected subjects on highly active antiretroviral therapy. AIDS 2002, 16, 63–73.
- Petrara, M.R.; Cattelan, A.M.; Zanchetta, M.; Sasset, L.; Freguja, R.; Gianesin, K.; Cecchetto, M.G.; Carmona, F.; De Rossi, A. Epstein-Barr virus load and immune activation in human immunodeficiency virus type 1-infected patients. J. Clin. Virol. 2012, 53, 195–200.
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18.
- Copeland, N.K.; Eller, M.A.; Kim, D.; Creegan, M.; Esber, A.; Eller, L.A.; Semwogerere, M.; Kibuuka, H.; Kiweewa, F.; Crowell, T.A.; et al. Brief Report: Increased Inflammation and Liver Disease in HIV/HBV-Coinfected Individuals. J. Acquir. Immune Defic. Syndr. 2021, 88, 310–313.
- Shmagel, K.V.; Saidakova, E.V.; Shmagel, N.G.; Korolevskaya, L.B.; Chereshnev, V.A.; Robinson, J.; Grivel, J.C.; Douek, D.C.; Margolis, L.; Anthony, D.D.; et al. Systemic inflammation and liver damage in HIV/hepatitis C virus coinfection. HIV Med. 2016, 17, 581–589.
- Chen, X.; Liu, X.; Duan, S.; Tang, R.; Zhou, S.; Ye, R.; Yang, Y.; Wang, J.; Yao, S.; He, N. Plasma Inflammatory Biomarkers Associated with Advanced Liver Fibrosis in HIV-HCV-Coinfected Individuals. Int. J. Environ. Res. Public Health 2020, 17, 9474.
- Borkow, G.; Bentwich, Z. Chronic immune activation associated with chronic helminthic and human immunodeficiency virus infections: Role of hyporesponsiveness and anergy. Clin. Microbiol. Rev. 2004, 17, 1012–1030.
- Brenchley, J.M.; Douek, D.C. Microbial Translocation Across the GI Tract. Annu. Rev. Immunol. 2012, 30, 149–173.
- Shan, L.; Siliciano, R.F. Unraveling the relationship between microbial translocation and systemic immune activation in HIV infection. J. Clin. Invest. 2014, 124, 2368–2371.
- Streeck, H.; Nixon, D.F. T cell immunity in acute HIV-1 infection. J. Infect. Dis. 2010, 202 (Suppl. 2), S302–S308.
- Guihot, A.; Dentone, C.; Assoumou, L.; Parizot, C.; Calin, R.; Seang, S.; Soulié, C.; Marcelin, A.G.; Calvez, V.; Autran, B.; et al. Residual immune activation in combined antiretroviral therapy-treated patients with maximally suppressed viremia. AIDS 2016, 30, 327–330.
- Méndez-Lagares, G.; Romero-Sánchez, M.C.; Ruiz-Mateos, E.; Genebat, M.; Ferrando-Martínez, S.; Muñoz-Fernández, M.Á.; Pacheco, Y.M.; Leal, M. Long- term suppressive combined antiretroviral treatment does not normalize the serum level of soluble CD14. J. Infect. Dis. 2013, 207, 1221–1225.
- Mudd, J.C.; Brenchley, J.M. Gut mucosal barrier dysfunction, microbial dysbiosis, and their role in HIV-1 disease progression. J. Infect. Dis. 2016, 214, S58–S66.
- Müller-Trutwin, M.; Hosmalin, A. Role for plasmacytoid dendritic cells in anti-HIV innate immunity. Immunol. Cell Biol. 2005, 83, 578–583.
- Grossman, Z.; Meier-Schellersheim, M.; Sousa, A.E.; Victorino, R.M.; Paul, W.E. CD4+ T-cell depletion in HIV infection: Are we closer to understanding the cause? Nat. Med. 2002, 8, 319–323.
- Cohen Stuart, J.W.; Hazebergh, M.D.; Hamann, D.; Otto, S.A.; Borleffs, J.C.; Miedema, F.; Boucher, C.A.; de Boer, R.J. The dominant source of CD4+ and CD8+ T-cell activation in HIV infection is antigenic stimulation. J. Acquir. Immune Defic. Syndr. 2000, 25, 203–211.
- Wolf, K.; Tsakiris, D.A.; Weber, R.; Erb, P.; Battegay, M.; Swiss HIV Cohort Study. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J. Infect. Dis. 2002, 185, 456–462.
- Vandergeeten, C.; Fromentin, R.; Chomont, N. The role of cytokines in the establishment, persistence and eradication of the HIV reservoir. Cytokine Growth Factor Rev. 2012, 23, 143–149.
- Catalfamo, M.; Le Saout, C.; Lane, H.C. The role of cytokines in the pathogenesis and treatment of HIV infection. Cytokine Growth Factor Rev. 2012, 23, 207–214.
- Keating, S.M.; Jacobs, E.S.; Norris, P.J. Soluble mediators of inflammation in HIV and their implications for therapeutics and vaccine development. Cytokine Growth Factor Rev. 2012, 23, 193–206.
- Kedzierska, K.; Crowe, S.M. Cytokines and HIV-1: Interactions and clinical implications. Antivir. Chem. Chemother. 2001, 12, 133–150.
- Kolte, L. Thymic function in HIV-infection. Dan. Med. J. 2013, 60, B4622.
- Bandera, A.; Ferrario, G.; Saresella, M.; Marventano, I.; Soria, A.; Zanini, F.; Sabbatini, F.; Airoldi, M.; Marchetti, G.; Franzetti, F.; et al. CD4+ T cell depletion, immune activation and increased production of regulatory T cells in the thymus of HIV-infected individuals. PLoS ONE 2010, 5, e10788.
- Ho Tsong Fang, R.; Colantonio, A.D.; Uittenbogaart, C.H. The role of the thymus in HIV infection: A 10 year perspective. AIDS 2008, 22, 171–184.
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522.
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875.
- Conway, J.M.; Perelson, A.S. Residual Viremia in Treated HIV+ Individuals. PLoS Comput. Biol. 2016, 12, e1004677.
- Luo, R.; Cardozo, E.F.; Piovoso, M.J.; Wu, H.; Buzón, M.J.; Martinez-Picado, J.; Zurakowski, R. Modeling HIV-1 2-LTR dynamics following raltegravir intensification. J. R. Soc. Interface 2013, 10, 20130186.
- Nou, E.; Lo, J.; Grinspoon, S.K. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS 2016, 30, 1495–1509.
- Deeks, S.G.; Phillips, A.N. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ 2009, 338, a3172.
- Iannetta, M.; Savinelli, S.; Rossi, R.; Mascia, C.; Marocco, R.; Vita, S.; Zuccalà, P.; Zingaropoli, M.A.; Mengoni, F.; Massetti, A.P.; et al. Myeloid and lymphoid activation markers in AIDS and non-AIDS presenters. Immunobiology 2019, 224, 231–241.
- Neuhaus, J.; Jacobs, D.R., Jr.; Baker, J.V.; Calmy, A.; Duprez, D.; La Rosa, A.; Kuller, L.H.; Pett, S.L.; Ristola, M.; Ross, M.J.; et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J. Infect. Dis. 2010, 201, 1788–1795.
- Hunt, P.W.; Martin, J.N.; Sinclair, E.; Bredt, B.; Hagos, E.; Lampiris, H.; Deeks, S.G. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 2003, 187, 1534–1543.
- Valdez, H.; Connick, E.; Smith, K.Y.; Lederman, M.M.; Bosch, R.J.; Kim, R.S.; St Clair, M.; Kuritzkes, D.R.; Kessler, H.; Fox, L.; et al. Limited immune restoration after 3 years’ suppression of HIV-1 replication in patients with moderately advanced disease. AIDS 2002, 16, 1859–1866.
- Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008, 5, e203.
- Tien, P.C.; Choi, A.I.; Zolopa, A.R.; Benson, C.; Tracy, R.; Scherzer, R.; Bacchetti, P.; Shlipak, M.; Grunfeld, C. Inflammation and mortality in HIV-infected adults: Analysis of the FRAM study cohort. J. Acquir. Immune Defic. Syndr. 2010, 55, 316–322.
- Andrade, B.B.; Hullsiek, K.H.; Boulware, D.R.; Rupert, A.; French, M.A.; Ruxrungtham, K.; Montes, M.L.; Price, H.; Barreiro, P.; Audsley, J.; et al. Biomarkers of inflammation and coagulation are associated with mortality and hepatitis flares in persons coinfected with HIV and hepatitis viruses. J. Infect. Dis. 2013, 207, 1379–1388.
- Sandler, N.G.; Wand, H.; Roque, A.; Law, M.; Nason, M.C.; Nixon, D.E.; Pedersen, C.; Ruxrungtham, K.; Lewin, S.R.; Emery, S.; et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 2011, 203, 780–790.
- Hunt, P.W.; Cao, H.L.; Muzoora, C.; Ssewanyana, I.; Bennett, J.; Emenyonu, N.; Kembabazi, A.; Neilands, T.B.; Bangsberg, D.R.; Deeks, S.G.; et al. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV- infected Ugandans initiating antiretroviral therapy. AIDS 2011, 25, 2123–2131.
- Duprez, D.A.; Neuhaus, J.; Kuller, L.H.; Tracy, R.; Belloso, W.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS ONE 2012, 7, e44454.
- Chun, T.W.; Murray, D.; Justement, J.S.; Hallahan, C.W.; Moir, S.; Kovacs, C.; Fauci, A.S. Relationship between residual plasma viremia and the size of HIV proviral DNA reservoirs in infected individuals receiving effective antiretroviral therapy. J. Infect. Dis. 2011, 204, 135–138.
- Ford, E.S.; Greenwald, J.H.; Richterman, A.G.; Rupert, A.; Dutcher, L.; Badralmaa, Y.; Natarajan, V.; Rehm, C.; Hadigan, C.; Sereti, I. Traditional risk factors and D-dimer predict incident cardiovascular disease events in chronic HIV infection. AIDS 2010, 24, 1509–1517.
- Borges, Á.H.; Silverberg, M.J.; Wentworth, D.; Grulich, A.E.; Fätkenheuer, G.; Mitsuyasu, R.; Tambussi, G.; Sabin, C.A.; Neaton, J.D.; Lundgren, J.D. Predicting risk of cancer during HIV infection: The role of inflammatory and coagulation biomarkers. AIDS 2013, 27, 1433–1441.
- McComsey, G.A.; Kitch, D.; Sax, P.E.; Tierney, C.; Jahed, N.C.; Melbourne, K.; Ha, B.; Brown, T.T.; Bloom, A.; Fedarko, N.; et al. Associations of inflammatory markers with AIDS and non-AIDS clinical events after initiation of antiretroviral therapy: AIDS clinical trials group A5224s, a substudy of ACTG A5202. J. Acquir. Immune Defic. Syndr. 2014, 65, 167–174.
- Jain, V.; Hartogensis, W.; Bacchetti, P.; Hunt, P.W.; Hatano, H.; Sinclair, E.; Epling, L.; Lee, T.H.; Busch, M.P.; McCune, J.M.; et al. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J. Infect. Dis. 2013, 208, 1202–1211.
- Ziegler, S.; Altfeld, M. Sex differences in HIV-1-mediated immunopathology. Curr. Opin. HIV AIDS 2016, 11, 209–215.
- Ballesteros-Zebadúa, P.; Villarreal, C.; Cocho, G.; Huerta, L.; Estrada, J.L. Differences in HIV-1 viral loads between male and female antiretroviral- untreated Mexican patients. Arch. Med. Res. 2013, 44, 296–301.
- Loupa, C.V.; Rodriguez, B.; McComsey, G.; Gripshover, B.; Salata, R.A.; Valdez, H.; Lisgaris, M.V.; Fulton, S.A.; Lederman, M.M. Gender differences in human immunodeficiency virus (HIV) RNA and CD4 cell counts among new entrants to HIV care. Clin. Microbiol. Infect. 2006, 12, 389–391.
- Collazos, J. Sexual dysfunction in the highly active antiretroviral therapy era. AIDS Rev. 2007, 9, 237–245.
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long- term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211.
- Fitch, K.V.; Srinivasa, S.; Abbara, S.; Burdo, T.H.; Williams, K.C.; Eneh, P.; Lo, J.; Grinspoon, S.K. Noncalcified coronary atherosclerotic plaque and immune activation in HIV-infected women. J. Infect. Dis. 2013, 208, 1737–1746.
- Yurkovetskiy, L.; Burrows, M.; Khan, A.A.; Graham, L.; Volchkov, P.; Becker, L.; Antonopoulos, D.; Umesaki, Y.; Chervonsky, A.V. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013, 39, 400–412.
- Seillet, C.; Laffont, S.; Trémollières, F.; Rouquié, N.; Ribot, C.; Arnal, J.F.; Douin- Echinard, V.; Gourdy, P.; Guéry, J.C. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor α signaling. Blood 2012, 119, 454–464.
- Libert, C.; Dejager, L.; Pinheiro, I. The X chromosome in immune functions: When a chromosome makes the difference. Nat. Rev. Immunol. 2010, 10, 594–604.
- Carrel, L.; Brown, C.J. When the Lyon(ized chromosome) roars: Ongoing expression from an inactive X chromosome. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160355.
- Mamrut, S.; Avidan, N.; Staun-Ram, E.; Ginzburg, E.; Truffault, F.; Berrih-Aknin, S.; Miller, A. Integrative analysis of methylome and transcriptome in human blood identifies extensive sex- and immune cell-specific differentially methylated regions. Epigenetics 2015, 10, 943–957.
- Butler, K.; Ritter, J.M.; Ellis, S.; Morris, M.R.; Hanson, D.L.; McNicholl, J.M.; Kersh, E.N. A Depot Medroxyprogesterone Acetate Dose That Models Human Use and Its Effect on Vaginal SHIV Acquisition Risk. J. Acquir. Immune Defic. Syndr. 2016, 72, 363–371.
- Prakash, M.; Kapembwa, M.S.; Gotch, F.; Patterson, S. Oral contraceptive use induces upregulation of the CCR5 chemokine receptor on CD4(+) T cells in the cervical epithelium of healthy women. J. Reprod. Immunol. 2002, 54, 117–131.
- Griesbeck, M.; Ziegler, S.; Laffont, S.; Smith, N.; Chauveau, L.; Tomezsko, P.; Sharei, A.; Kourjian, G.; Porichis, F.; Hart, M.; et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-α Production in Women. J. Immunol. 2015, 195, 5327–5336.
- Mellors, J.W.; Margolick, J.B.; Phair, J.P.; Rinaldo, C.R.; Detels, R.; Jacobson, L.P.; Muñoz, A. Prognostic value of HIV-1 RNA, CD4 cell count, and CD4 Cell count slope for progression to AIDS and death in untreated HIV-1 infection. JAMA 2007, 297, 2349–2350.
More