Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Diana Amantea and Version 2 by Camila Xu.
Tumor necrosis factor (TNF)-α-stimulated gene 6 (TSG-6), the first soluble chemokine-binding protein to be identified in mammals, inhibits chemotaxis and transendothelial migration of neutrophils and attenuates the inflammatory response of dendritic cells, macrophages, monocytes, and T cells. This immunoregulatory protein is a pivotal mediator of the therapeutic efficacy of mesenchymal stem/stromal cells (MSC) in diverse pathological conditions, including neuroinflammation.
- neurodegeneration
- neuroinflammation
- stroke
- TSG-6
1. Biological Effects of TSG-6
Among the multitude of genes regulated by TNF-α is the TNF-α-induced protein 6 (TNFAIP6) gene, coding for the secretory protein TSG-6, initially identified in human fibroblasts lines and peripheral blood mononuclear cells [1][2][85,86]. TSG-6 can be induced by diverse cytokines (i.e., TNF, IL-1) and other inflammatory stimuli (i.e., LPS) and is a member of the family of hyaluronate (HA) binding proteins involved in cell–cell and cell–matrix interactions during inflammation and tumorigenesis [1][3][4][85,87,88]. In fact, although constitutively expressed in some tissues, including the brain and spinal cord, TSG-6 is generally upregulated in response to inflammation in a wide variety of cell types, such as monocytes/macrophages, dendritic cells, astrocytes, mesenchymal stem/stromal cells (MSCs), vascular smooth muscle cells (VSMCs), and fibroblasts [5][6][7][8][9][89,90,91,92,93]. TSG-6 has a variety of activities, including the modulation of immune and stromal cells function and the regulation of extracellular matrix organization and interaction with cell surface receptors and soluble mediators (e.g., chemokines) (figure in Section 4 of original contextSection 4) [10][94]. The anti-inflammatory functions of TSG-6 rely on direct modulation of inflammatory cells and on the regulation of the assembly/organization of HA matrices that underlie its immunosuppressive properties [11][12][13][14][15][16][17][18][95,96,97,98,99,100,101,102]. TSG-6 was also shown to inhibit NETs release from bone-marrow-derived neutrophils [19][103]. Moreover, the original evidence that TSG-6 produced by MSCs underlies their immunomodulatory and reparative functions [20][21][104,105] has stimulated interest in understanding the role of this molecule in a number of pathological conditions. Indeed, an increasing body of experimental evidence suggests that the main function of this protein is to protect tissue from the damaging effects caused by inflammation, as demonstrated in animal models of acute myocardial infarction [20][104], atherosclerosis [22][106], lung injury [23][24][107,108], arthritis [25][26][27][28][29][109,110,111,112,113], spinal cord injury [30][114], and, most notably, acute brain injury [31][32][33][34][35][36][115,116,117,118,119,120].
2. Immunomodulatory Functions of TSG-6 In Vitro
TSG-6 was the first soluble chemokine-binding protein to be identified in mammals, as it binds to a wide range of chemokines belonging to both CC and CXC subfamilies, hindering their presentation and interaction with matrix molecules [37][38][121,122]. By interacting with CXCL8, TSG-6 blocks its binding to endothelial heparan sulphate (HS), a mechanism implicated in its potent (in vitro and in vivo) inhibitory activity on neutrophil chemotaxis and transendothelial migration (Figure 1) [37][38][39][40][41][121,122,123,124,125]. The latter mechanism has been reported to underlie the protective effects of TSG-6 in a number of degenerative and inflammatory experimental conditions, including acute brain injury [20][21][33][42][43][44][104,105,117,126,127,128]. Moreover, binding to other chemokines that can be recognized by diverse leukocytes (e.g., CCR5, CCR7, and CXCR4) strongly suggests that TSG-6 may regulate recruitment and migration of different white blood cells (Figure 1) [37][38][121,122]. Accordingly, TSG-6 was reported to regulate the function of dendritic cells, macrophages, monocytes, and T cells attenuating their inflammatory responses and promoting immune tolerance [12][20][23][43][45][46][47][48][49][96,104,107,127,129,130,131,132,133].
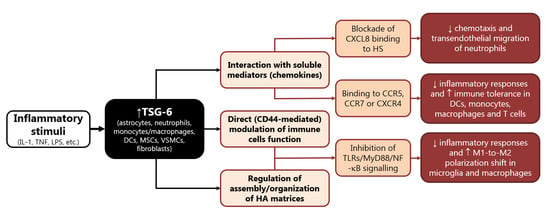
Figure 1. Main mechanisms involved in the immunomodulatory functions of TSG-6. Abbreviations: DC: dendritic cells, HA: hyaluronate, HS: heparan sulphate, IL: interleukin, LPS: lipopolysaccharide, MSCs: mesenchymal stem/stromal cells, myeloid differentiation primary response: MyD, TLRs: Toll-like receptors, NF-κB: nuclear factor kappa B, TNF: tumor necrosis factor, TSG: tumor necrosis factor-α-stimulated gene, VSMCs: vascular smooth muscle cells.
In human neutrophils, TSG-6 is constitutively present in the secretory lactoferrin-positive granules; monocytes, macrophages, myeloid dendritic cells, and neutrophils produce high levels of TSG-6 in response to inflammatory triggers (LPS or cytokines) [50][134]. Conversely, anti-inflammatory cytokines, namely IL-4 and IL-10, dampen LPS-induced TSG-6 expression in human leukocytes [50][134].
In turn, TSG-6 acts in an autocrine mode on macrophages to promote their transition from proinflammatory toward anti-inflammatory phenotypes (Figure 1) [23][49][51][107,133,135]. In particular, this immunomodulatory protein inhibits the association between TLR-4 and MyD88, thus suppressing the activation of NF-κB, and prevents the expression of pro-inflammatory mediators (e.g., IL-6, TNF-α, IL-1β and inducible nitric oxide synthase, iNOS, STAT1, and STAT3), while elevating the expression of anti-inflammatory proteins (e.g., CD206, IL-4, and IL-10) [12][14][23][45][52][53][96,98,107,129,136,137].
The majority of the studies performed to date on TSG-6 were focused on its ability to mediate the therapeutic efficacy of MSC in a number of pathological conditions, including neuroinflammation, since it plays a major role in the modulation of sterile inflammation [10][54][94,138]. Bone MSCs stimulated with TNF-α release TSG-6 that has been reported to inhibit production of inflammatory cytokines triggered by LPS in cultured rat astrocytes [31][55][115,139]. This effect was mediated by inhibition of NF-κB signaling pathway and was suggested to underlie the protective role of bone NSCs on BBB damage induced by intracerebral or subarachnoid hemorrhage in rat [31][55][115,139].
Similarly, MSCs and adipose-derived stem cells (ADSCs) exposed to TNF-α increased their expression of TSG-6 that, once secreted, inhibited the release of inflammatory mediators (i.e., IL-1β, IL-6, TNF-α, iNOS) by cultured BV2 microglia [56][57][140,141]. This effect was shown to be mediated by the interaction of TSG-6 with CD44 and subsequent blockade of NF-κB inflammatory signaling in BV2 microglia [57][141]. In fact, TSG-6 binds and forms stable complexes with HA, thus stabilizing its interaction with the membrane receptor CD44, and resulting in negative regulation of TLR-4-dependent signaling [11][13][58][59][95,97,142,143]. Accordingly, TSG-6 downregulated the TLR2/MyD88/NF-κB signaling (Figure 1) and reduced the production of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, in primary microglia treated with a specific TLR2 agonist, an effect that was suggested to underlie the ability of TSG-6 to attenuate neuropathic pain caused by chronic constriction injury in rats [60][144]. A similar mechanism was described in cultured murine macrophages, whereby TSG-6 released by stimulated MSCs downregulated TLR2-mediated nuclear translocation of NF-κB, by binding CD44 directly or through a complex with HA [14][98]. The ability of TSG-6 to inhibit proliferation and release of inflammatory mediators was also suggested to occur through reduced activation of p38 and JNK signaling in rat macrophages [61][62][145,146].
TSG-6 also suppressed LPS-induced TNF-α production and the expression of the inflammatory M1 phenotype in human monocyte-derived macrophages [63][147], while it prevented NF-κB activation by inhibiting the association of MyD88 with TLR4 in mouse macrophages (Figure 1) [23][107]. Interestingly, TLR2-related pathways were reported to play a major role in the regulation of the polarization state of microglia, while NF-κB and p38 were implicated in polarization of microglia/macrophages during ischemic stroke injury [64][65][66][67][68][69][148,149,150,151,152,153]. This strongly supports the hypothesis that TSG-6 may affect M1 vs. M2 polarization shift of myeloid cells under neuroinflammatory conditions. In line with this speculation is the evidence that BV2 microglia treated with MSCs displayed reduced LPS-induced expression of early and late markers of M1 phenotype (iNOS, IL-1β, CD16, CD86), and concomitant elevation of typical markers of M2 polarization (CD206, Arg1) [70][71][154,155]. Notably, most of these effects were lost when LPS-primed BV2 microglia were exposed to MSCs in which TSG-6 was knocked down [70][154]. In this context, TSG-6 was suggested to modulate microglia polarization shift by preventing LPS-induced phosphorylation of STAT3 [70][154]. The inhibitory effect of TSG-6 on STAT3 phosphorylation was also demonstrated to underlie polarization toward M2 phenotype in murine macrophages, thus underlying protection in mice undergone LPS-induced inflammatory lung injury or liver inflammation due to alcoholic hepatitis [23][72][107,156].
3. TSG-6 as Mediator of the Neuroprotective Functions of MSCs
The anti-inflammatory and immunoregulatory functions of TSG-6 have been studied in a number of neurological disorders (Figure 2), although most studies mainly focused on the role of this multifunctional protein in the protective effects of MSCs, while little is known on its direct mechanisms and on its functions when released from endogenous sources.
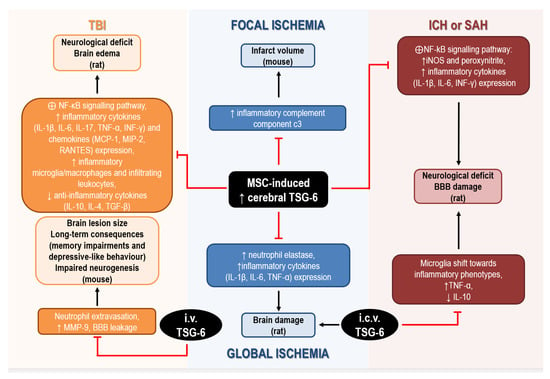
Figure 2. Mechanisms underlying the neuroprotective effects of TSG-6 in rodents subjected to acute brain injury. Abbreviations: BBB: blood–brain barrier, IL: interleukin, INF: interferon, MCP: monocyte chemoattractant protein, MIP: macrophage inflammatory protein, MMP: matrix metalloprotease, MSC: mesenchymal stem/stromal cells, TGF: transforming growth factor, TNF: tumor necrosis factor, TSG: tumor necrosis factor-α-stimulated gene.
In global cerebral ischemia caused by cardiac arrest in rat, intravenous administration of MSCs significantly reduced brain damage by upregulating cerebral expression of TSG-6, while attenuating the ischemia-induced elevation of neutrophil elastase and of inflammatory cytokines (i.e., IL-1β, IL-6, and TNF-α) expression [35][73][119,157]. Although administration of recombinant TSG-6 in the lateral ventricle was effective in reducing histological and functional deficits associated with global ischemia, these authors did not investigate the mechanisms involved in cerebral upregulation of TSG-6 by MSCs, but they hypothesized that TSG-6 was actually released by MSCs migrating to the brain [35][119]. In fact, intravenous infusion of MSCs, in which TSG-6 expression was silenced by siRNA, failed to attenuate brain inflammation in ischemic rats [73][157].
Interestingly, bone marrow-derived MSCs are effective in ameliorating stroke outcomes and, despite limited knowledge of their exact neuroprotective mechanisms, their immune-suppressive and anti-inflammatory effects are likely to play a crucial role. This is supported by the evidence that intravenous injection of MSCs in mice reduced the elevation of brain and blood levels of the inflammatory complement component C3 induced by focal cerebral ischemia, thus resulting in reduced infarct volume [74][158]. These effects were also associated with elevation of brain and blood levels of TSG-6, 8 h after MSCs administration, strongly suggesting its involvement in their protective effects [74][158].
The ability of TSG-6 to attenuate neuroinflammatory reactions was also demonstrated in rats exposed to collagenase to induce intracerebral hemorrhage (ICH), intravenously transplanted with MSCs. In this model, the protective effects of MSCs on BBB and their anti-inflammatory effects were ascribed to the increased mRNA and protein expression of TSG-6 detected in the brain 24 h and 48 h after ICH [75][159]. In particular, although not directly demonstrated, TSG-6 elevation was suggested to underlie suppression of the activation of NF-kB signaling pathway and downstream reduction of iNOS and peroxynitrite levels [75][159]. More recent studies have confirmed the ability of bone MSCs to reduce neurological deficits and BBB damage caused by ICH or SAH in rat, through TSG-6 release, since these protective effects were abolished after silencing TSG-6 by siRNA [31][55][115,139]. In particular, Tang et al. (2021) speculated that TSG-6, likely secreted via exosomes by bone MSCs, acts through a paracrine mechanism to regulate activated astrocytes to preserve BBB integrity. In addition, TSG-6 was demonstrated to mediate the anti-inflammatory effects of bone MSCs, by inhibiting NF-κB signaling pathways and the cerebral increase of inflammatory cytokines (e.g., IL-1β, IL-6, INF-γ) and peroxynitrite caused by ICH or SAH in rats [31][55][115,139].
Enhanced expression of TSG-6 and downstream suppression of NF-κB signaling pathways have also been suggested to underlie the anti-inflammatory effects of MSCs or NS309 (a small conductance Ca2+-activated K+ channels activator) in rodent models of traumatic brain injury (TBI). In fact, intravenous MSCs transplantation 2 h after TBI, or i.p. administration of NS309 30 min before TBI, caused upregulation of TSG-6 in the injured cortex up to 72 h after the insult in rat [36][76][120,160]. This was associated with reduced levels of inflammatory cytokines (i.e., IL-1β, IL-6, IL-17, TNF-α, INF-γ) and chemokines (i.e., MCP-1, MIP-2, RANTES), lower density of inflammatory microglia/macrophages and peripheral infiltrating leukocytes, together with elevated levels of anti-inflammatory cytokines (i.e., IL-10, IL-4, TGF-β1) in the lesioned tissue [36][76][120,160]. Notably, knockdown of TSG-6 using in vivo transfection with TSG-6 specific shRNA partially reversed the protective and anti-inflammatory effects of NS309 against TBI [76][160]. To further support its beneficial effects, there is the evidence that intravenous treatment with TSG-6 decreased neutrophil extravasation, matrix metalloproteinase (MMP)-9 expression and the resulting BBB leakage caused by TBI in mice [33][117]. Remarkably, acute administration of TSG-6 within 24 h of TBI not only reduced brain lesion size at two weeks, but it also promoted neurogenesis and attenuated long-term consequences of TBI, such as memory impairments and depressive-like behavior [33][117].