Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 4 by Conner Chen.
The dysregulation of RasGRP1 (Ras guanine nucleotide-releasing proteins 1(RasGRPs)) is known to contribute to numerous disorders that range from autoimmune and inflammatory diseases and schizophrenia to neoplasia. Given its position at the crossroad of cell development, inflammation, and cancer, RASGRP1 has garnered interest from numerous disciplines.
- Ras
- RasGEF
- RasGRP1
1. Ras Guanine Nucleotide Exchange Factors: Introduction
Ras guanine nucleotide exchange factors (RasGEFs) are composed of three families of proteins: Ras guanine nucleotide-releasing proteins (RasGRPs), Son of Sevenless (SOS), and Ras guanine nucleotide-releasing factors (RasGRFs). The RasGRP family consists of four members, RasGRP1, RasGRP2, RasGRP3, and RasGRP4, the SOS family is composed of two members, SOS1 and SOS2, and the RasGRF family is also composed of two members, RasGRF1 and RasGRF2. The commonality is that they catalyze the removal of guanosine diphosphate (GDP) from GTPases, such as Ras and Rap, and allow for its replacement [1] (Figure 1). While Ras itself possesses intrinsic GTPase and guanine nucleotide exchange activities, the basal activity is low. The activation of the canonical Ras pathway is characterized by the phosphorylation of Raf, Mek, and Erk. The active GTP (guanosine triphosphate)-bound Ras has a wide range of downstream effects at the cellular level, such as a proliferation, differentiation, and apoptosis. Given these fundamental roles, numerous disease processes have been attributed to the dysregulation of Ras and RasGEFs, which range from autoimmune and inflammatory diseases to neoplasia.
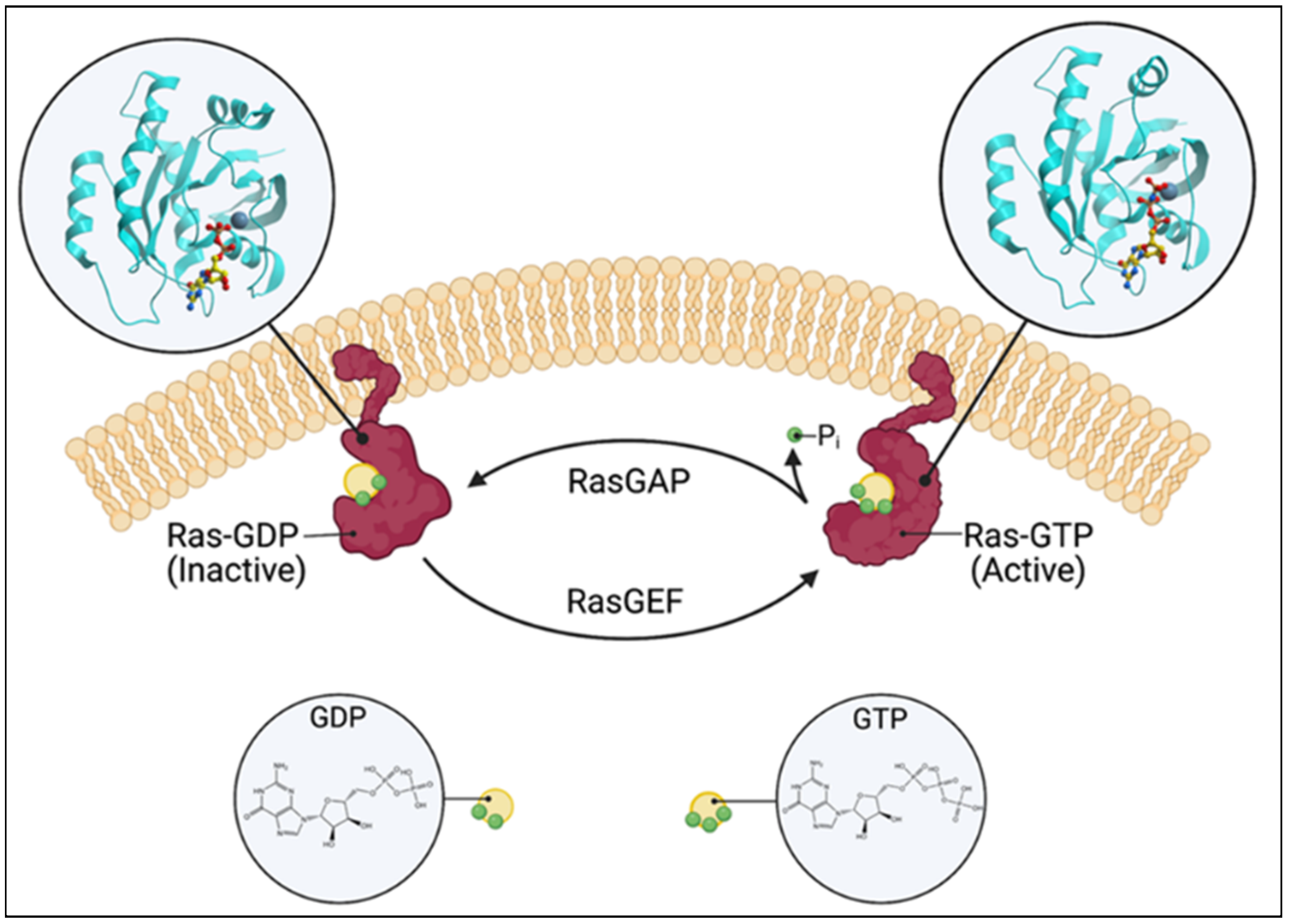
Figure 1. Schematic of the Ras switching cycle. Ras cycles between the GTP-bound active state and the GDP-bound inactive state. RasGAP catalyzes the hydrolysis of GTP, and RasGEF facilitates guanine nucleotide exchange. Created with BioRender.com.
2. RasGRP1: Role in Cancer
2.1. Lymphoma and Leukemia
While loss-of-function RasGRP1 mutants have been described in humans [2][3][4], no oncogenic mutant of RasGRP1 has been identified. These loss-of-function RasGRP1 mutants lead to the development of autoimmune lymphoproliferative syndrome (ALPS), CD4+ T cell lymphopenia, recurrent infections, hepatosplenomegaly, and lymphadenopathy [2][3][4]. It is important to note that some patients with loss-of-function RasGRP1 mutants develop Epstein–Barr virus (EBV)-induced B cell lymphoma. However, studies have found RasGRP1 to be overexpressed in nearly half of all T cell acute lymphoblastic leukemias (T-ALL) [5][6]. Retroviral insertion studies in mice have also identified wild-type RasGRP1 as a leukemogenic oncogene [7][8][9]. Furthermore, the dysregulation of RasGRP1 in mice and cell lines has been shown to lead to the development of thymic lymphomas and T cell leukemias [6][10][11]. Interestingly, cell lines with a high RasGRP1 expression required a cocktail of IL-2 (interleukin (IL)), -7, and -9 for proliferation [6][11]. Additionally, leukemia driven by the overexpression of RasGRP1 and K-RasG12D are mutually exclusive and represent the distinct mechanisms of leukemogenesis [6]. This is consistent with the finding from a later study that identified RasGRP1 as a negative regulator of Ras signaling in Kras−/− NrasQ61R/+-driven leukemia [12]. Various studies have shown that the dysregulation of RasGRP1 itself is insufficient for leukemogenesis [6][13]; however, it does bestow a proliferative advantage in bone marrow progenitors over wild type cells [13]. Consistent with Knudson’s “two-hit” theory that was proposed over 50 years ago [14], the dysregulation of RasGRP1 requires a second cooperating oncogene or cytokine stimulation for transformation [5][6][11]. The knockout of RasGRP1 negative regulators has also been shown to be oncogenic; specifically, DGKα−/− DGKζ−/− (diacylglycerol kinase (DGK)) double knockout mice develop thymic lymphoma due to the failure to prevent the overactivation of RasGRP1 and Ras [15]. Beyond the role of RasGRP1 as an oncogene, its overexpression has been documented to be a mechanism of resistance to MEK inhibitors [16].
No RasGRP1-specific small molecule inhibitors currently exist. Since the overexpression of RasGRP1 renders T-ALL cells responsive to pro-tumorigenic cytokines [11], PI3K inhibitors have been tested as a monotherapy in mice, but with no success [17]. Others have tried to target the RasGRP1/Ras/Erk pathway in T cell lymphoblastic lymphomas (T-LBL), which are morphologically and immunophenotypically identical to T-ALL [18]. Bromodomain-containing protein 2 (BRD2) binds to the promotor region of Rasgrp1 and conveys a doxorubicin resistance in some T-LBL patients [19]. The targeting of BRD2 via a bromodomain and extra-terminal (BET) inhibitor improved the therapeutic efficacy in vitro and in a patient-derived xenograft mouse model [19]. Diacylglycerol (DAG) and its analogues have long been known to activate RasGRP1 in T and B cells [20][21], and the treatment of B cell lymphoma-derived cell lines with DAG analogues promoted apoptosis [21][22]. This proapoptotic pathway induced by DAG analogues is mediated by the protein kinase C (PKC)/RasGRP1/Erk pathway [21][22].
2.2. Squamous Cell Carcinoma
While studying the role of RasGRP1 in skin tumors, one group found that the overexpression of RasGRP1, driven by a K5 promotor, in keratinocytes resulted in the development of spontaneous skin tumors [23][24]. These tumors were mostly benign papillomas and there were lesser numbers of squamous cell carcinomas. Due to the observation that the incidence of tumors development was higher in co-housed animals, it was hypothesized that wounding contributed to tumor development. Indeed, when RasGRP1-K5 transgenic mice were subjected to full-thickness incision wounding, 50% of them developed skin tumors [24]. The proposed mechanism is that the act of wounding caused the release of the granulocyte colony-stimulating factor (G-CSF) by keratinocytes [23][24], and G-CSF acted in an autocrine and paracrine fashion to cooperate with RasGRP1 in the development of skin tumors [25]. When the same RasGRP1-K5 transgenic mice were subjected to multistage skin carcinogenesis protocol, 7,12-dimethylbenz(a)anthracene (DBMA) as carcinogen, and 12-O-tetradecanoylphorbol-13-acetate (TPA) as tumor promoters, it was found that the squamous cell carcinomas that developed in the transgenic mice were larger, less differentiated, and more invasive [26]. Additionally, the overexpression of RasGRP1 was found to partially replace the DMBA induction [26]. Conversely, RasGRP1 knockout mice have impaired skin tumorigenesis, evidenced by a reduced epidermal hyperplasia induced by TPA [27][28]. To study other coopering mechanisms of oncogenesis in keratinocytes, one group transduced keratinocytes derived from a Li-Fraumeni patient with RasGRP1 and found that the keratinocytes acquired morphologic changes that are associated with a transformation [29]. This result supports the idea that RasGRP1 cooperates with other genes because patients with Li-Fraumeni syndrome are deficient in p53, a well-known tumor suppressor gene.
2.3. Colorectal Cancer
Surprisingly, RasGRP1 acts as a tumor suppressor in colonic epithelium; furthermore, RasGRP1 can be used as a biomarker for predicting the efficacy of anti-epidermal growth factor receptor (EGFR) therapy for CRC (colorectal cancer) patients [30][31]. The RasGRP1 expression levels decrease with the progression of CRC and predict the poor clinical outcome of patients [31]. Mechanistically, the same group found that RasGRP1 suppresses the proliferation of the KRas mutant and negatively regulates the EGFR/SOS1/Ras signal in CRC cells [31]. This mechanism may explain its tumor suppressor activity in colorectal cancer in contrast to its oncogenic activity in most other neoplasias.
2.4. Hepatocellular Carcinoma
RasGRP1 has been found to be upregulated in hepatocellular carcinomas (HCC) [32]; furthermore, a high RasGRP1 expression is associated with the tumor size, tumor–node–metastasis (TNM) stage, and Barcelona Clinic Liver Cancer stage [32]. At the cellular level, in Huh7 and PLC cells, the downregulation of RasGRP1 inhibited cell proliferation, whereas the overexpression of RasGRP1 promoted cell proliferation [32]. Specific protein 1 (Sp1) was identified to bind the Rasgrp1 promotor and is a positive regulator [32].
2.5. Breast Cancer
The role of RasGRP1 in breast cancer has only recently been studied. Specifically, it was found that the upregulation of Rasgrp1 was associated with an improved overall survival in breast cancer [33], as well as overall survival and disease-free survival in the triple-negative breast cancer subtype [33][34]. The molecular mechanism that underlies these observations is unknown.References
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279.
- Mao, H.; Yang, W.; Latour, S.; Yang, J.; Winter, S.; Zheng, J.; Ni, K.; Lv, M.; Liu, C.; Huang, H.; et al. RASGRP1 mutation in autoimmune lymphoproliferative syndrome-like disease. J. Allergy Clin. Immunol. 2018, 142, 595–604.e516.
- Somekh, I.; Marquardt, B.; Liu, Y.; Rohlfs, M.; Hollizeck, S.; Karakukcu, M.; Unal, E.; Yilmaz, E.; Patiroglu, T.; Cansever, M.; et al. Novel Mutations in RASGRP1 are Associated with Immunodeficiency, Immune Dysregulation, and EBV-Induced Lymphoma. J. Clin. Immunol. 2018, 38, 699–710.
- Winter, S.; Martin, E.; Boutboul, D.; Lenoir, C.; Boudjemaa, S.; Petit, A.; Picard, C.; Fischer, A.; Leverger, G.; Latour, S. Loss of RASGRP1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol. Med. 2018, 10, 188–199.
- Oki, T.; Kitaura, J.; Watanabe-Okochi, N.; Nishimura, K.; Maehara, A.; Uchida, T.; Komeno, Y.; Nakahara, F.; Harada, Y.; Sonoki, T.; et al. Aberrant expression of RasGRP1 cooperates with gain-of-function NOTCH1 mutations in T-cell leukemogenesis. Leukemia 2012, 26, 1038–1045.
- Hartzell, C.; Ksionda, O.; Lemmens, E.; Coakley, K.; Yang, M.; Dail, M.; Harvey, R.C.; Govern, C.; Bakker, J.; Lenstra, T.L.; et al. Dysregulated RasGRP1 responds to cytokine receptor input in T cell leukemogenesis. Sci. Signal. 2013, 6, ra21.
- Kim, R.; Trubetskoy, A.; Suzuki, T.; Jenkins, N.A.; Copeland, N.G.; Lenz, J. Genome-based identification of cancer genes by proviral tagging in mouse retrovirus-induced T-cell lymphomas. J. Virol. 2003, 77, 2056–2062.
- Dupuy, A.J.; Morgan, K.; von Lintig, F.C.; Shen, H.; Acar, H.; Hasz, D.E.; Jenkins, N.A.; Copeland, N.G.; Boss, G.R.; Largaespada, D.A. Activation of the Rap1 guanine nucleotide exchange gene, CalDAG-GEF I, in BXH-2 murine myeloid leukemia. J. Biol. Chem. 2001, 276, 11804–11811.
- Suzuki, T.; Shen, H.; Akagi, K.; Morse, H.C.; Malley, J.D.; Naiman, D.Q.; Jenkins, N.A.; Copeland, N.G. New genes involved in cancer identified by retroviral tagging. Nat. Genet. 2002, 32, 166–174.
- Klinger, M.B.; Guilbault, B.; Goulding, R.E.; Kay, R.J. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis independently of T-cell receptors. Oncogene 2005, 24, 2695–2704.
- Ksionda, O.; Melton, A.A.; Bache, J.; Tenhagen, M.; Bakker, J.; Harvey, R.; Winter, S.S.; Rubio, I.; Roose, J.P. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene 2016, 35, 3658–3668.
- Wen, Z.; Yun, G.; Hebert, A.; Kong, G.; Ranheim, E.A.; Finn, R.; Rajagoplan, A.; Li, S.; Zhou, Y.; Yu, M.; et al. Nras Q61R/+ and Kras-/- cooperate to downregulate Rasgrp1 and promote lympho-myeloid leukemia in early T-cell precursors. Blood 2021, 137, 3259–3271.
- Karra, L.; Romero-Moya, D.; Ksionda, O.; Krush, M.; Gu, Z.; Mues, M.; Depeille, P.; Mullighan, C.; Roose, J.P. Increased baseline RASGRP1 signals enhance stem cell fitness during native hematopoiesis. Oncogene 2020, 39, 6920–6934.
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823.
- Guo, R.; Wan, C.K.; Carpenter, J.H.; Mousallem, T.; Boustany, R.M.; Kuan, C.T.; Burks, A.W.; Zhong, X.P. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc. Natl. Acad. Sci. USA 2008, 105, 11909–11914.
- Lauchle, J.O.; Kim, D.; Le, D.T.; Akagi, K.; Crone, M.; Krisman, K.; Warner, K.; Bonifas, J.M.; Li, Q.; Coakley, K.M.; et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature 2009, 461, 411–414.
- Ksionda, O.; Mues, M.; Wandler, A.M.; Donker, L.; Tenhagen, M.; Jun, J.; Ducker, G.S.; Matlawska-Wasowska, K.; Shannon, K.; Shokat, K.M.; et al. Comprehensive analysis of T cell leukemia signals reveals heterogeneity in the PI3 kinase-Akt pathway and limitations of PI3 kinase inhibitors as monotherapy. PLoS ONE 2018, 13, e0193849.
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951.
- Tian, X.P.; Cai, J.; Ma, S.Y.; Fang, Y.; Huang, H.Q.; Lin, T.Y.; Rao, H.L.; Li, M.; Xia, Z.J.; Kang, T.B.; et al. BRD2 induces drug resistance through activation of the RasGRP1/Ras/ERK signaling pathway in adult T-cell lymphoblastic lymphoma. Cancer Commun. 2020, 40, 245–259.
- Stone, J.C.; Stang, S.L.; Zheng, Y.; Dower, N.A.; Brenner, S.E.; Baryza, J.L.; Wender, P.A. Synthetic bryostatin analogues activate the RasGRP1 signaling pathway. J. Med. Chem. 2004, 47, 6638–6644.
- Stang, S.L.; Lopez-Campistrous, A.; Song, X.; Dower, N.A.; Blumberg, P.M.; Wender, P.A.; Stone, J.C. A proapoptotic signaling pathway involving RasGRP, Erk, and Bim in B cells. Exp. Hematol. 2009, 37, 122–134.
- Leo, I.R.; Aswad, L.; Stahl, M.; Kunold, E.; Post, F.; Erkers, T.; Struyf, N.; Mermelekas, G.; Joshi, R.N.; Gracia-Villacampa, E.; et al. Integrative multi-omics and drug response profiling of childhood acute lymphoblastic leukemia cell lines. Nat. Commun. 2022, 13, 1691.
- Oki-Idouchi, C.E.; Lorenzo, P.S. Transgenic overexpression of RasGRP1 in mouse epidermis results in spontaneous tumors of the skin. Cancer Res. 2007, 67, 276–280.
- Diez, F.R.; Garrido, A.A.; Sharma, A.; Luke, C.T.; Stone, J.C.; Dower, N.A.; Cline, J.M.; Lorenzo, P.S. RasGRP1 transgenic mice develop cutaneous squamous cell carcinomas in response to skin wounding: Potential role of granulocyte colony-stimulating factor. Am. J. Pathol. 2009, 175, 392–399.
- Obermueller, E.; Vosseler, S.; Fusenig, N.E.; Mueller, M.M. Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res. 2004, 64, 7801–7812.
- Luke, C.T.; Oki-Idouchi, C.E.; Cline, J.M.; Lorenzo, P.S. RasGRP1 overexpression in the epidermis of transgenic mice contributes to tumor progression during multistage skin carcinogenesis. Cancer Res. 2007, 67, 10190–10197.
- Sharma, A.; Fonseca, L.L.; Rajani, C.; Yanagida, J.K.; Endo, Y.; Cline, J.M.; Stone, J.C.; Ji, J.; Ramos, J.W.; Lorenzo, P.S. Targeted deletion of RasGRP1 impairs skin tumorigenesis. Carcinogenesis 2014, 35, 1084–1091.
- Sharma, A.; Luke, C.T.; Dower, N.A.; Stone, J.C.; Lorenzo, P.S. RasGRP1 is essential for ras activation by the tumor promoter 12-O-tetradecanoylphorbol-13-acetate in epidermal keratinocytes. J. Biol. Chem. 2010, 285, 15724–15730.
- Fonseca, L.L.; Yang, W.S.; Geerts, D.; Turkson, J.; Ji, J.; Ramos, J.W. RasGRP1 induces autophagy and transformation-associated changes in primary human keratinocytes. Transl. Oncol. 2021, 14, 100880.
- Gbenedio, O.M.; Bonnans, C.; Grun, D.; Wang, C.Y.; Hatch, A.J.; Mahoney, M.R.; Barras, D.; Matli, M.; Miao, Y.; Garcia, K.C.; et al. RasGRP1 is a potential biomarker to stratify anti-EGFR therapy response in colorectal cancer. JCI Insight 2019, 5, 127552.
- Depeille, P.; Henricks, L.M.; van de Ven, R.A.; Lemmens, E.; Wang, C.Y.; Matli, M.; Werb, Z.; Haigis, K.M.; Donner, D.; Warren, R.; et al. RasGRP1 opposes proliferative EGFR-SOS1-Ras signals and restricts intestinal epithelial cell growth. Nat. Cell Biol. 2015, 17, 804–815.
- Zhang, X.; Zhuang, H.; Han, F.; Shao, X.; Liu, Y.; Ma, X.; Wang, Z.; Qiang, Z.; Li, Y. Sp1-regulated transcription of RasGRP1 promotes hepatocellular carcinoma (HCC) proliferation. Liver Int. 2018, 38, 2006–2017.
- Wang, S.; Beeghly-Fadiel, A.; Cai, Q.; Cai, H.; Guo, X.; Shi, L.; Wu, J.; Ye, F.; Qiu, Q.; Zheng, Y.; et al. Gene expression in triple-negative breast cancer in relation to survival. Breast Cancer Res. Treat. 2018, 171, 199–207.
- Chou, C.W.; Huang, Y.M.; Chang, Y.J.; Huang, C.Y.; Hung, C.S. Identified the novel resistant biomarkers for taxane-based therapy for triple-negative breast cancer. Int. J. Med. Sci. 2021, 18, 2521–2531.
More