Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 3 by Conner Chen.
The bone marrow is responsible for providing the body with a constant supply of millions of circulating blood cells and platelets. Haematopoietic stem/progenitor cells (HSPCs) are rare, self-renewing, multipotent progenitors that produce all types of these blood cells via haematopoiesis. Maintaining these HSPCs requires key signals and interactions with both haematopoietic and non-haematopoietic cells within the bone marrow microenvironment. Understanding these cellular communications may provide insight into haematological diseases and uncover better strategies for treatment.
- bone marrow microenvironment
- Haematopoietic stem/progenitor cells (HSPCs)
- Endosteal Niche
- Perivascular Niche
1. Introduction
The bone marrow is a heterogeneous environment comprised of various haematopoietic and non-haematopoietic cells, including osteoblasts and osteoclasts, mesenchymal stromal cells (MSCs), neurons, immune cells, adipocytes, sinusoidal endothelium and perivascular stromal cells (Figure 1). Haematopoietic stem/progenitor cells (HSPCs) reside in haematopoietic niches of the bone marrow, and this supportive microenvironment is essential for the long-term maintenance of a stable pool of HSPCs. HSPC functions, including proliferation, quiescence, adhesion and differentiation, are regulated by these non-haematopoietic cells through the release of variety of factors [1][2].
The haematopoietic niche is anatomically divided into two compartments: the internal endosteal bone surface and the associated perivascular network of blood vessels (Figure 1). These niches are closely related to the vascular structures, arterioles and sinusoids, respectively, and influence HSPC function in a variety of different ways [3].
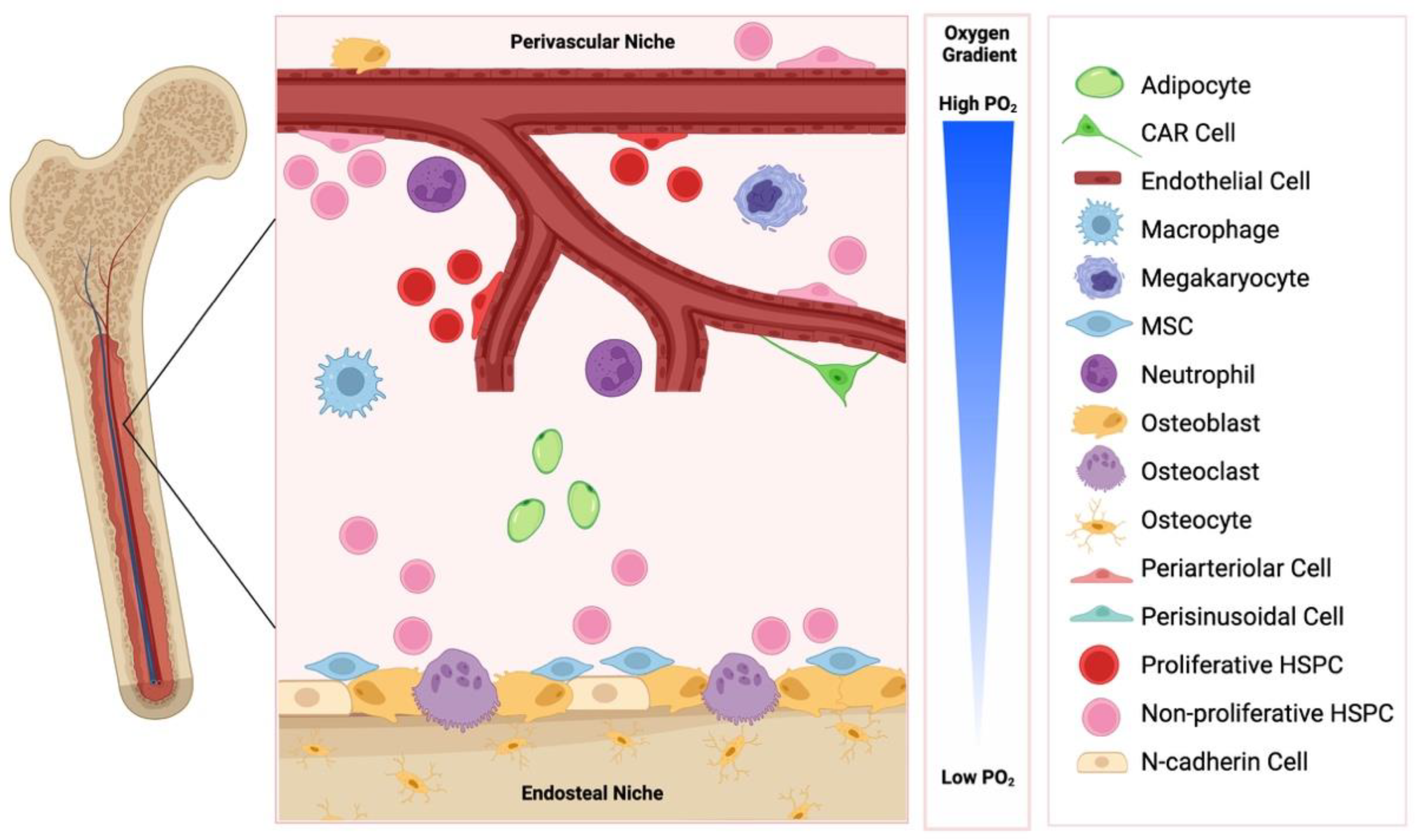
Figure 1. The endosteal and perivascular bone marrow niche. These niches are comprised of distinct cell types, are influence haematopoietic stem/progenitor cell (HSPC) function in a variety of ways. CAR: CXCL12 abundant reticular cells; MSC: mesenchymal stem cell. Created with BioRender.com.
2. The Endosteal Niche
The endosteal niche is defined anatomically by close proximity to cortical or trabecular bone and has a high content of osteoblasts and osteoclasts (Figure 1). In addition to their primary functions of bone remodelling, osteoblasts and osteoclasts have been implicated in regulating HSPC function, lodgement, and egress from the bone marrow. For example, osteoblastic lineage cells have been shown to control HSPC regulation in vivo via parathyroid hormone (PTH) receptor dependent signalling [4], and osteoclasts have been implicated in stem cell mobilisation in an osteopontin (OPN), stem cell factor (SCF, also known as KITL) and C-X-C motif chemokine 12 (CXCL12)/stromal cell derived factor 1 (SDF-1) mediated manner [5]. Additionally, components of the extracellular matrix, including OPN, the seven-transmembrane-spanning calcium sensing receptor (CaSR) or the sympathetic nervous system can impact HSPC function [6][7][8][9]. OPN negatively regulates HSPC numbers, and OPN−/− mice exhibited increased numbers of stem cells, reduced primitive haematopoietic cell apoptosis and enhanced HSPC cycling [7][8]. Additionally, maintenance of HSPCs in a quiescent state is promoted by the interaction between angiopoietin-1 on osteoblasts and the receptor tyrosine kinase Tie2 on HSPC [10]. UDGP-galactose ceramide galactosyltransferase-deficient (Cgt−/−) mice exhibited aberrant nerve conduction and displayed no HSPC egress from the bone marrow following granulocyte colony-stimulating factor (G-CSF) or fucoidan administration, due to downregulated CXCL12 expression [9]. The Ca2+ content of the niche, mediated via CaSR, dictated the localisation of HSPCs, and CaSR deficient HSPCs were normal in number, proliferative and differentiation function migration and homing, however, they exhibited defective localisation, due to defective adhesion to collagen [6]. Adhesion molecules are involved in HSPC retention within the bone marrow, and as observed with CaSR deficient HSPCs, are important for directing the correct localisation of HSPCs within the bone marrow niche. While several adhesion factors have been well characterised in this function, the role of the cell adhesion molecule, N-cadherin, in HSPC lodgement into endosteal niches remains controversial. Whilst initial work demonstrated that HSPC expressed N-cadherin [11], subsequent studies have brought this into question [12]. Conditional deletion of the N-cadherin gene from osteoblasts and haematopoietic cells does not alter the frequency or the number of HSPC in the bone marrow, or their long-term or serial reconstitution potential [13]. By contrast, expression of a dominant-negative mutant of N-cadherin that inhibits both homotypic and heterotypic interactions of N-cadherin in donor HSPC reduced endosteal lodgement and compromised long-term engraftment, whereas overexpression of a wild-type N-cadherin increased endosteal lodgement and self-renewal ability [14]. Overexpression of short hairpin RNA (shRNA) specific for silencing N-cadherin gene expression in HSPC, increased HSPC proliferation, and reduced long-term engraftment and HSPC lodgement to endosteal surfaces [15]. Taken together, these studies demonstrate that the expression of N-cadherin on HSPC and its role in HSPC lodgement and function requires further clarification. The bone marrow niche itself provides a privileged environment that supports HSPC self-renewal [16][17]. When bone marrow cells are injected into tissue engineered ectopic ossicles or in the circulation following lethal irradiation of mice to eliminate host HSPC, donor HSPCs colonised and self-renewed within the ossicles, and reconstituted haematopoiesis. This process was mediated by the proto-oncogene c-myc [17]. C-myc has been shown to control the balance between HSPC self-renewal and differentiation [18]. Conditional deletion of c-myc in haematopoietic cells enhanced HSPC self-renewal but inhibited differentiation and exhibited increased expression of adhesion molecules. Further, these c-myc deficient HSPC can home to and lodge into endosteal niches but failed to differentiate into mature leukocytes. Conversely, overexpression of c-myc compromised HSPC reconstitution potential following lethal BM irradiation in mouse model recipients [18]. Taken together, these studies demonstrate the importance of c-myc in normal HSPC differentiation and function and suggest that c-myc can mediate the interaction between HSPCs and the bone marrow niche. Perhaps not surprisingly, c-myc, located at 8q24, is one of the most frequently activated genes in acute myeloid leukaemia (AML) and overexpression plays an important role in leukaemogenesis. In addition to supporting HSPC self-renewal, the bone marrow microenvironment supports differentiation of haematopoietic progenitor cells into various lineages through the regulation of various signalling pathways, particularly the canonical Wnt signalling pathway. β-catenin-deficient bone marrow microenvironment maintained HSPCs but exhibited a decreased capacity to support primitive haematopoietic cells, correlated with decreased osteoblasts and production of FGF, SCF, and VCAM-1 [19]. These findings highlight the importance of the canonical Wnt signalling pathway in the bone marrow microenvironment for the maintenance of haematopoiesis. Osteoblasts and osteoclasts have also been shown to be required for B cell development. Osteoblast ablation results in a rapid decrease in the numbers of pre-pro-B and pro-B cells [20]. In contrast, inhibition of osteoclast function results in relocalisation of B cell progenitors to the spleen [21]. Taken together, these studies suggest that the endosteal surface is required for B lymphopoiesis.3. The Perivascular Niche
The perivascular niche is defined anatomically by close proximity to sinusoidal vascular endothelium, including surrounding supportive elements such as extracellular matrix and stromal cells (Figure 1). The blood vessels of the bone marrow are separate from the peripheral circulation [22]. It is well established that homing to the bone marrow involves an initial ‘capture’ step whereby circulating HSPC directly interact with the bone marrow endothelium. The sinusoidal endothelial cells of the bone marrow constitutively express adhesion molecules, including P-selectin, E-selectin, and vascular cell adhesion molecule-1 (VCAM-1) [23][24], which are believed to facilitate this ‘capture’ step. It is therefore unsurprising that HSPCs primarily reside in the perivascular niche in the bone marrow and spleen, however, some HSPCs are associated with endosteum [25][26]. As such, the perivascular niche is currently believed to help maintain primitive HSPCs in an undifferentiated state, and several studies have shown that the perivascular niche provides biomolecular signals that can influence HSPC function, implicating the perivascular secretome in influencing the fate of HSPCs [27]. Adventitial reticular cells, which express high levels of CXCL12/SDF-1 (CAR: CXCL12 adventitial reticular cells), and are of presumed mesenchymal origin, can alter stem cell function. Targeted deletion of CXCR4, the ligand for CXCL12/SDF-1, decreased HSPC numbers and increased sensitivity to myelotoxic injury, without impairing expansion of the more mature progenitor cells [28], highlighting that the interaction between CXCR4 and CXCL12/SDF-1 is important for the maintenance of the HSPC quiescent pool. CXCL12/SDF-1−/− embryos have reduced HSPC number and function, which can be overcome by enforced CXCL12/SDF-1 expression in vascular endothelial cells [29], highlighting the importance of this chemokine in this process. Other growth factors have also been implicated in HSPC function in the bone marrow. Deletion of vascular endothelial growth factor receptor 2 (VEGFR2) in adult mice blocked regeneration of bone marrow sinusoidal endothelial cells and prevented haematopoietic reconstitution [30]. Additionally, VCAM-1 and very late antigen 4 (VLA-4) adhesion molecules are implicated in the localisation and adhesion of HSPCs within the bone marrow niche [31][32]. Interestingly, CXCL12 expression and HSPC retention in the bone marrow is regulated by the sympathetic nervous system, most likely by sympathetic nerve fibres that synapse on perivascular cells around a subset of blood vessels, which thereby regulate CXCL12 expression and HSPC mobilisation via circadian oscillations [9][33]. Several other signalling pathways have been implicated in the proliferation and self-renewal in vivo. For example, wingless (Wnt) signalling is activated and necessary in the bone marrow niche to limit HSPC proliferation and preserve reconstituting capacity, and Dickkopf-1 expression in osteoblast cells reduces in vivo repopulating ability and quiescence [34]. Several subsets of bone marrow cells have been implicated in supporting immune cell function (reviewed in [35][36]). CAR cells have been shown to create a niche for HSPCs and immune cells produced in the bone marrow. Structurally, CAR cells possess long processes, and HSPCs, plasma cells, natural killer cells, plasmacytoid dendritic cells, and B cell precursors have been identified to be in contact with these processes [37][38][39][40]. Another key niche component that maintains HSPCs and is primarily expressed by perivascular cells throughout the bone marrow is SCF. Whilst HSPC frequency and function were not affected by conditional deletion of Scf from haematopoietic cells or osteoblasts, deletion from endothelial or leptin receptor-expressing (LepR) perivascular stromal cells led to HSPC depletion [41], highlighting the importance of SCF in promoting HSPC maintenance in the perivascular niche. By contrast, conditional deletion of SCF from LepR+ endothelial cells led to depleted common myeloid progenitors (CMPs), common lymphoid progenitors (CLPs), granulocyte-macrophage progenitors (GMPs), megakaryocyte-erythrocyte progenitors (MEPs), pre-MEPs, and colony-forming units-erythroid (CFU-E), as well as erythroid and myeloid blood cells. Importantly, this was not a result of HSPC depletion [42]. Taken together, this reveals cellular specialisation within the perivascular niche that is perhaps mediated in a LepR-dependent manner. An additional key physiological regulator of HSPC function within the perivascular niche are oxygen tensions. Compared to the atmospheric oxygen tension of 21%, the HSPC niche exhibits low oxygen tensions in the range of 1–6% oxygen [43], well below the 2–9% considered by some scientists to be cellular normoxia [44]. Hypoxia activates a range of molecular responses that maintain HSPCs in a quiescent and pluripotent state, while HSPCs residing in close proximity to the vascular niche actively cycle and replenish circulating cells [45].References
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S.A. The bone marrow microenvironment—Home of the leukemic blasts. Blood Rev. 2017, 31, 277–286.
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell. Biol. 2017, 19, 214–223.
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643.
- Calvi, L.M. Osteoblastic activation in the hematopoietic stem cell niche. Ann. N. Y. Acad. Sci. 2006, 1068, 477–488.
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664.
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006, 439, 599–603.
- Stier, S.; Ko, Y.; Forkert, R.; Lutz, C.; Neuhaus, T.; Grunewald, E.; Cheng, T.; Dombkowski, D.; Calvi, L.M.; Rittling, S.R.; et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791.
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239.
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421.
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161.
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841.
- Kiel, M.J.; Radice, G.L.; Morrison, S.J. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell 2007, 1, 204–217.
- Kiel, M.J.; Acar, M.; Radice, G.L.; Morrison, S.J. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell Stem Cell 2009, 4, 170–179.
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Iwasaki, H.; Hembree, M.; Yin, T.; Nakamura, Y.; Gomei, Y.; Takubo, K.; Shiama, H.; et al. Cadherin-based adhesion is a potential target for niche manipulation to protect hematopoietic stem cells in adult bone marrow. Cell Stem Cell 2010, 6, 194–198.
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Iwasaki, H.; Nakamura, Y.; Gomei, Y.; Suda, T. Knockdown of N-cadherin suppresses the long-term engraftment of hematopoietic stem cells. Blood 2010, 116, 554–563.
- Schneider, A.; Taboas, J.M.; McCauley, L.K.; Krebsbach, P.H. Skeletal homeostasis in tissue-engineered bone. J. Orthop. Res. 2003, 21, 859–864.
- Song, J.; Kiel, M.J.; Wang, Z.; Wang, J.; Taichman, R.S.; Morrison, S.J.; Krebsbach, P.H. An in vivo model to study and manipulate the hematopoietic stem cell niche. Blood 2010, 115, 2592–2600.
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763.
- Nemeth, M.J.; Mak, K.K.; Yang, Y.; Bodine, D.M. beta-Catenin expression in the bone marrow microenvironment is required for long-term maintenance of primitive hematopoietic cells. Stem Cells 2009, 27, 1109–1119.
- Zhu, J.; Garrett, R.; Jung, Y.; Zhang, Y.; Kim, N.; Wang, J.; Joe, G.J.; Hexner, E.; Choi, Y.; Taichman, R.S.; et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 2007, 109, 3706–3712.
- Mansour, A.; Anginot, A.; Mancini, S.J.; Schiff, C.; Carle, G.F.; Wakkach, A.; Blin-Wakkach, C. Osteoclast activity modulates B-cell development in the bone marrow. Cell Res. 2011, 21, 1102–1115.
- Tamma, R.; Ribatti, D. Bone Niches, Hematopoietic Stem Cells, and Vessel Formation. Int. J. Mol. Sci. 2017, 18, 151.
- Schweitzer, K.M.; Drager, A.M.; van der Valk, P.; Thijsen, S.F.; Zevenbergen, A.; Theijsmeijer, A.P.; van der Schoot, C.E.; Langenhuijsen, M.M. Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am. J. Pathol. 1996, 148, 165–175.
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973.
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121.
- Chen, J.Y.; Miyanishi, M.; Wang, S.K.; Yamazaki, S.; Sinha, R.; Kao, K.S.; Seita, J.; Sahoo, D.; Nakauchi, H.; Weissman, I.L. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 2016, 530, 223–227.
- Barnhouse, V.; Petrikas, N.; Crosby, C.; Zoldan, J.; Harley, B. Perivascular Secretome Influences Hematopoietic Stem Cell Maintenance in a Gelatin Hydrogel. Ann. Biomed. Eng. 2021, 49, 780–792.
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988.
- Ara, T.; Tokoyoda, K.; Sugiyama, T.; Egawa, T.; Kawabata, K.; Nagasawa, T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity 2003, 19, 257–267.
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009, 4, 263–274.
- Williams, D.A.; Rios, M.; Stephens, C.; Patel, V.P. Fibronectin and VLA-4 in haematopoietic stem cell-microenvironment interactions. Nature 1991, 352, 438–441.
- Hao, J.; Zhou, H.; Nemes, K.; Yen, D.; Zhao, W.; Bramlett, C.; Wang, B.; Lu, R.; Shen, K. Membrane-bound SCF and VCAM-1 synergistically regulate the morphology of hematopoietic stem cells. J. Cell Biol. 2021, 220, e202010118.
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447.
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283.
- Mercier, F.E.; Ragu, C.; Scadden, D.T. The bone marrow at the crossroads of blood and immunity. Nat. Rev. Immunol. 2011, 12, 49–60.
- Weber, J.M.; Calvi, L.M. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone 2010, 46, 281–285.
- Kohara, H.; Omatsu, Y.; Sugiyama, T.; Noda, M.; Fujii, N.; Nagasawa, T. Development of plasmacytoid dendritic cells in bone marrow stromal cell niches requires CXCL12-CXCR4 chemokine signaling. Blood 2007, 110, 4153–4160.
- Noda, M.; Omatsu, Y.; Sugiyama, T.; Oishi, S.; Fujii, N.; Nagasawa, T. CXCL12-CXCR4 chemokine signaling is essential for NK-cell development in adult mice. Blood 2011, 117, 451–458.
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.I.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718.
- Sugiyama, T.; Nagasawa, T. Bone marrow niches for hematopoietic stem cells and immune cells. Inflamm. Allergy Drug Targets 2012, 11, 201–206.
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462.
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell 2019, 24, 477–486.e476.
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161.
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell. Biol. 2008, 9, 285–296.
- Winkler, I.G.; Barbier, V.; Wadley, R.; Zannettino, A.C.; Williams, S.; Levesque, J.P. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: Serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood 2010, 116, 375–385.
More