Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Jose M Gonzalez-Granado.
Inflammatory bowel disease (IBD) encompasses a group of heterogeneous diseases that entail chronic, relapsing gastrointestinal tract inflammation of inexactly known etiology and pathogenesis. IBD is clinically classified as Crohn’s disease (CD) or ulcerative colitis (UC) based on symptoms, disease location, and histopathological characteristics.
- innate immune system
- inflammatory bowel disease
- ulcerative colitis
- Crohn's disease
1. Introduction
Inflammatory bowel disease (IBD) encompasses a group of heterogeneous diseases that entail chronic, relapsing gastrointestinal tract inflammation of inexactly known etiology and pathogenesis. IBD etiology may involve the host immune system, genetic variability, and environmental factors [1]. IBD is clinically classified as Crohn’s disease (CD) or ulcerative colitis (UC) based on symptoms, disease location, and histopathological characteristics. UC causes long-lasting inflammation and superficial ulcerative disease in the colon, whereas CD is a transmural disease often associated with granuloma formation and can appear in any part of the gastrointestinal tract [2,3,4,5][2][3][4][5]. IBD can be associated with life-threatening conditions, including primary sclerosing cholangitis, blood clots, and colon cancer [6]. IBD is usually diagnosed between the ages of 20 and 40 years, but can start at any age. IBD shows alternating periods of clinical relapse and remission.
The intestinal mucosa is composed of epithelial cells, goblet and Paneth cells, stroma and immune cells. The intestinal epithelium includes a monolayer of epithelial cells closely bound by tight junctions and interposed with immune cells. The intestine is structured as a series of protrusions known as villi and invaginations called crypts of Lieberkühn [7]. The epithelium participates in nutrient absorption and, at the same time, interposes a physical barrier to the contents of the intestinal lumen. The epithelium also interacts with the intestinal microbiota and the immune system, sending receiving signals to and from both.
The epithelium includes goblet and Paneth cells, which, respectively, produce mucus and antimicrobial peptides, thus limiting the spread of luminal microorganisms [7]. A marked reduction in goblet cell numbers has been linked to a loss of mucus layer thickness in Crohn’s disease [8], and abnormal mucus composition has been reported in UC [9]. Beneath the epithelium, the lamina propria contains stromal cells, including fibroblasts, myofibroblasts, and perivascular pericytes. These cells serve functions in fibrosis and wound healing [7], and may be related to the aggravation of UC through their capacity to produce chemokines, including chemokine (C-C motif) ligand (CCL)19, CCL21, and the immune-system regulator interleukin (IL)-33 [10].
Plasma cells release immunoglobulin (Ig)A, inhibiting the infiltration of pathogenic microorganisms and helping to sustain a homeostatic equilibrium between the host and commensal microbiota. Both the epithelium and other non-immune intestinal components are important mediators of intestinal homeostasis and IBD pathophysiology, reviewed in [11,12][11][12]. However, some of the functions of these non-immune cells are mediated through interaction with components of the immune system, as will be described in this revisewarch.
The immune system confers host defense against pathogens and provides anti-tumor protection. At the same time, regulatory mechanisms counterbalance these responses to prevent reactions against self and innocuous external antigens, thus promoting a state of tolerance.
The immune system can be classified into innate and adaptive immunity. Innate immunity, composed of myeloid cells among other elements, initiates rapid and nonspecific responses to conserved structural motifs on microorganisms. Innate immune cells (IIC) express pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and Nod-like receptors (NLR), allowing them to distinguish pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). IIC promote host defense and inflammation by generating cytokines and chemokines, triggering the complement cascade and phagocytosis, or stimulating adaptive immunity by presenting antigens. Notable IIC include neutrophils, monocytes, macrophages, and dendritic cells (DCs) [13,14][13][14].
2. IBD Pathophysiology
The etiology of IBD remains elusive, but IBD appears to be sustained in genetically susceptible individuals by an impaired immune response against intestinal microorganisms. This abnormal immune response is associated with dysregulation of both innate and adaptive immune responses.
IBD features breach the epithelial barrier of specific zones in the intestine, and non-resolving mucosal damage is thought to be an important characteristic of the disease [15]. While generally unknown, the cause of this damage could be related to an infectious agent [16], a chemical compound [1], or a metabolic alteration probably related to diet-mediated dysbiosis [17]. The disease is then thought to be perpetuated by deficient resolution of the inflammatory response to this initial injury [18]. Unsuccessful resolution of inflammation is possibly supported by disruption of tolerance towards commensal microorganisms or autologous signals of tissue damage [15,19][15][19].
There is also some uncertainty as to whether the epithelial barrier alterations precede or follow the development of inflammation in the lamina propria [20].
2.1. Innate Immune Cells in the Pathogenesis of IBD
In IBD, the innate immune system is the first responder to PAMPs and to molecules released from damaged or dying cells, known as DAMPs. DAMPs and PAMPs activate the innate immune system by interacting with PRRs. These patterns can be sensed by several components of the innate immune system, including granulocytes, neutrophils, monocytes, myeloid-derived suppressor cells, macrophages, and dendritic cells. In addition, these patterns can also be recognized by non-immune cells, such as intestinal epithelial cells (IECs) and myofibroblasts.
IICs respond to these signals, temporarily enhance the epithelial barrier, and clean up the effects of inflammation [21]. The implication of IIC in IBD is the focus of this revisewarch.
2.1.1. Neutrophils in Gut Homeostasis
Neutrophils are the most numerous immune cells in the human circulation and are quickly recruited to sites of infection or inflammation [22], forming the first line of immune defense.
When the intestinal barrier is damaged, neutrophils are recruited from the circulation to the inflamed tissue through a plethora chemotactic gradients formed by cytokines such as IL-1β, IL-6, and tumor necrosis factor (TNF)-α; chemokines such as CCL8, chemokine (C-X-C motif) ligand (CXCL)10, and macrophage inflammatory protein 2 (MIP)-2 (also known as CXCL2); and growth factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) [23,24,25][23][24][25]. Neutrophil recruitment is also mediated by bacteria-derived molecules such as formyl-methionyl-leucyl-phenylalanine and short-chain fatty acids (SCFAs) [26,27,28][26][27][28] (Figure 1).
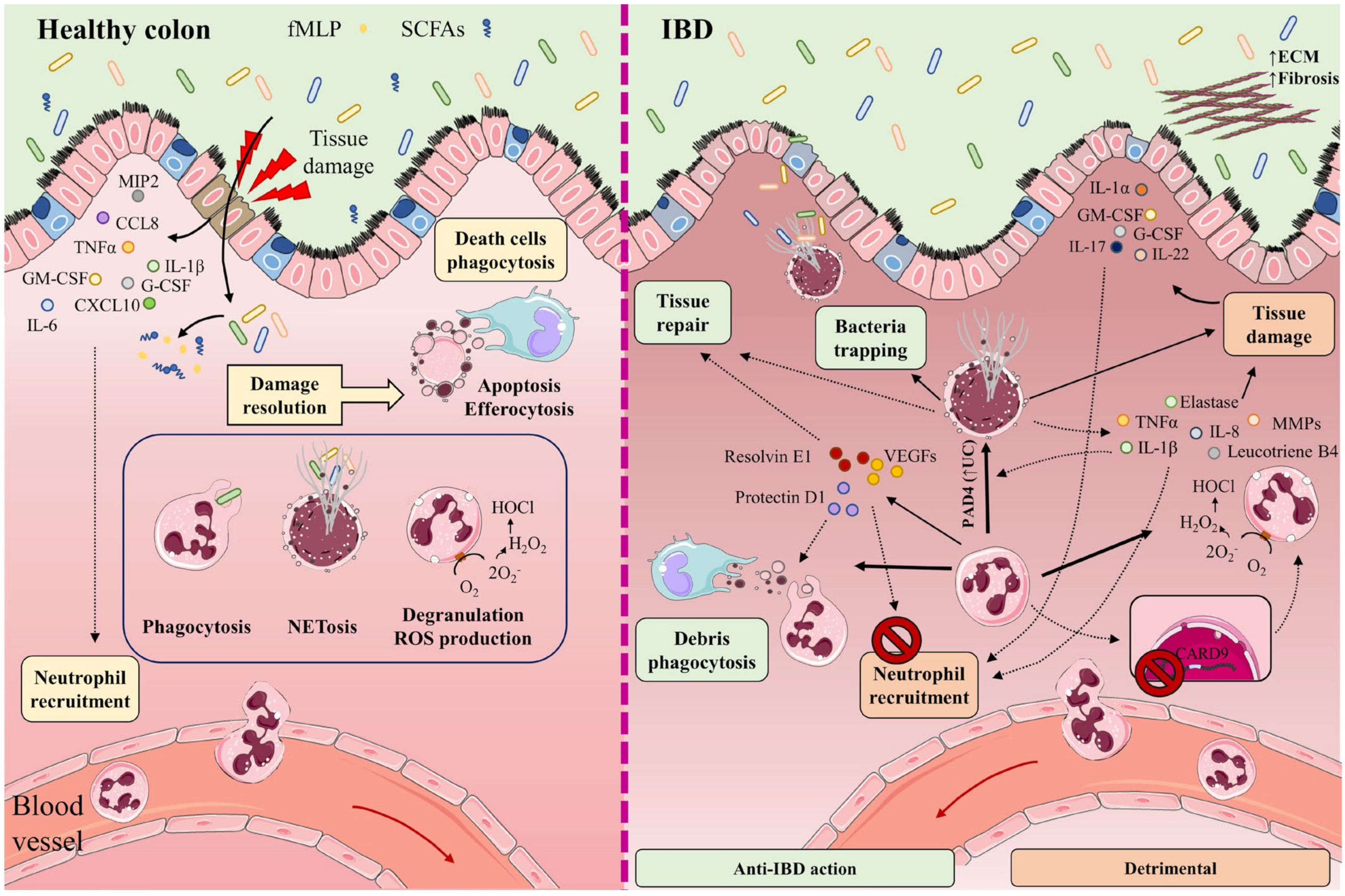
Figure 1. In healthy intestine (left), damage to the intestinal barrier triggers the recruitment of neutrophils from the circulation to the inflamed tissue along a chemotactic gradient formed by cytokines (IL-1β, IL-6, TNF-α), chemokines (CCL8, CXCL10, MIP-2), and growth factors (GM-CSF, G-CSF). Neutrophil recruitment is also mediated by bacteria-derived molecules such as formyl-methionyl-leucyl-phenylalanine (fMLP) and short-chain fatty acids (SCFAs). The recruited neutrophils participate in the elimination of microorganisms through phagocytosis, degranulation, reactive oxygen species (ROS) generation, and the release of neutrophil extracellular traps (NETs). Once their functions are completed, neutrophils undergo apoptosis and efferocytosis, facilitating the resolution of inflammation, tissue repair, and a return to normal tissue homeostasis. The participation of neutrophils and NETs in IBD is a double-edged sword (right). Neutrophils cooperate in wound healing and the resolution of inflammation by releasing vascular endothelial growth factors (VEGFs) and lipid mediators (protectin D1, resolvin E1). These factors impede neutrophil recruitment and promote phagocytosis. NETs impede the spread of microorganisms by trapping them in an environment of microbicidal components and stimulate the healing of the intestinal mucosa. Neutrophils directly cause tissue damage by releasing neutrophil elastase, proteases (MMPs), pro-inflammatory cytokines (IL-8, TNF-α, IL-1β), leukotriene B4, and ROS. These factors provoke not only injury to the epithelial barrier, but also the recruitment of neutrophils and other immune cells to the inflamed tissue. Neutrophil recruitment is also promoted by the cytokines IL-1α, IL-17, IL-22, G-CSF, and GM-CSF. Lack of the IBD protective gene CARD9 in neutrophils enhances ROS generation. IL-8, TNF-α, and PAD4 (increased in UC patients) contribute to NET production. Accumulation of NETs in the colon is accompanied by the induction of tissue damage and inflammation, as NETs also boost TNF-α and IL-1β production. Part of the figure was generated by using pictures from Servier Medical Art.
Neutrophils participate in the elimination of microorganisms through phagocytosis, degranulation, the generation of reactive oxygen species (ROS), and the release of neutrophil extracellular traps (NETs). NETs are mesh-like structures made of DNA and its histone scaffold together with granule components such as myeloperoxidase (MPO), cathepsin G, neutrophil elastase, and protease 3. NETs protrude from the membrane of the activated neutrophil to restrain large microorganisms, activate complement factors, and therefore facilitate contained lysis through their bactericidal and permeability-increasing actions [29,30,31,32][29][30][31][32]. Once their functions are completed, neutrophils undergo apoptosis and efferocytosis, facilitating resolution of the inflammatory response, repair, and a return to normal tissue homeostasis [33,34,35][33][34][35].
2.1.2. Neutrophils in the Gut during IBD
Some studies have described the participation of NETs in IBD as a double-edged sword. On the one hand, NETs can impede the spread of microorganisms by trapping them in an environment of microbicidal components, while also stimulating the healing of the intestinal mucosa upon injury and helping to sustain the stability of the intestinal epithelium [36,37][36][37]. On the other hand, increased neutrophil activity and exacerbated NETs production can impair intestinal mucosal barrier function, damage the intestinal epithelium, and accentuate disease symptoms [38]. Neutrophils directly promote tissue damage by releasing proteases such as matrix metalloproteinases (MMPs) and neutrophil elastase, and by altering membrane properties by releasing ROS [39] (Figure 1).
Proteomics studies and microscopy validations have identified eleven neutrophil- and NETs-associated proteins with increased abundance in biopsies from UC patients [40], and similar results have been reported by others [39].
Dextran sodium sulfate (DSS)-induced colitis in mice promotes the accumulation of NETs in the colon, accompanied by the induction of epithelial cell death by apoptosis, breakage of tight junctions, increased permeability, and augmented bacterial translocation and inflammation [41]. Moreover, NETs accumulation boosts TNF-α and IL-1β production in plasma by signaling via the ERK1/2 pathway. The reduction of NETs protects against colitis and inhibits the augmentation of pro-inflammatory factors implicated in IBD [41].
An increase in neutrophil activity has been observed in IBD patients [42]. This increase is associated with the release of TNF-α and the presence of lipopolysaccharides, two factors that contribute to neutrophil activation. Other factors that can contribute to NETs production are IL-8 produced by endothelial cells, and an increased abundance of protein arginine deiminase 4 (PAD4) [30,31,43][30][31][43]. PAD4 is more abundant in intestinal tissue from UC patients than healthy individuals [31], and in damaged tissue rather than healthy tissue from the same individual [30,31,32][30][31][32]. PAD4 mediates histone citrullination, an important event in NETosis that precedes chromatin decondensation and DNA release.
An analysis of NETs-associated proteins in colon samples from patients with UC, CD, and colon cancer showed a greater abundance of PAD4, MPO, neutrophil elastase, and citrullination histone H3 (CitH3) in UC than in CD. Moreover, neutrophils in UC tissues produced more NETs upon treatment with TNF-α [44]. In a similar way, increased expression of Ly6G, CitH3, and PAD4 has been found in mouse colon in a model of colitis induced by 2,4,6-trinitrobenzene sulfonic acid (TNBS) [45]. This effect was associated with damage to the intestinal epithelial barrier [45]. Reduced NETs formation ameliorated colitis symptoms and tissue damage [46,47,48,49][46][47][48][49].
Neutrophils promote IBD gut inflammation by producing high levels of ROS that impair the epithelial barrier and promote redox-sensitive inflammatory pathways [50]. The epithelial barrier is also damaged by neutrophil-produced proteases, pro-inflammatory cytokines such as IL-8, TNF-α, and leukotriene B4, which additionally recruit monocytes and more neutrophils to the inflamed tissue [51,52][51][52].
Another cytokine recently proposed to promote neutrophil recruitment to colonic tissue is IL-22, whose levels correlate with neutrophil infiltration [53]. Neutrophils can be activated by the cytokines IL-1β and IL-18 [54], produced during inflammasome assembly [55[55][56],56], and upon the release by necrotic cells of the nuclear ‘alarmin’ IL-1α [57]. Other mediators of neutrophil action altered in IBD include GM-CSF and G-CSF [58] and IL-17A and IL-17F [59], which act through the IL-23–IL-17A–G-CSF axis [60[60][61],61], providing a possible explanation for the continuous regeneration of neutrophils in IBD [15]. Neutrophils also express the IBD-protective gene caspase recruitment domain 9 (Card9), which provides them with the capacity to protect against DSS-induced colitis; a lack of CARD9 enhances mitochondrial dysfunction and ROS generation, leading to neutrophil apoptosis and increased inflammation [62].
The intestinal epithelium constitutes a physical barrier that isolates subepithelial tissues from luminal contents, providing a fundamental support for intestinal homeostasis. Neutrophils are associated with bystander tissue damage, but also play a role in epithelial restitution [63].
As well as killing microorganisms, neutrophils cooperate in wound healing and the resolution of inflammation by releasing vascular endothelial growth factors and beneficial lipid mediators such as protectin D1 and resolvin E1. These factors impede further neutrophil recruitment and augment the phagocytosis of apoptotic neutrophils by macrophages. Neutrophils also remove cellular debris from sites of inflammation by phagocytosis [61,64,65,66][61][64][65][66]. The release of proteases within NETs can regulate cytokine function by proteolysis [67], and this phenomenon might also regulate cytokines in IBD.
Contrasting these protective effects, the accumulation of hyperactivated neutrophils promotes an alteration of crypt structure and the formation crypt abscesses. This process features a disproportionate enzymatic reaction, generation of the pro-inflammatory cytokines TNF-α and IL-1β, and the secretion of the non-cytokine inflammatory molecules α defensins and calprotectin, attracting monocytes, T cells, and more neutrophils to the inflammation site and promoting the pathogenesis of IBD [68,69,70,71,72][68][69][70][71][72]. Neutrophils can also promote goblet-cell depletion, a main feature of IBD [73,74,75][73][74][75].
Neutrophils are also, themselves, regulated by the effects of intestinal epithelial cells during intestinal inflammation, a topic recently reviewed in [63].
In summary, neutrophils play intricate roles in intestinal inflammation, contributing to the elimination of invading pathogens and epithelial restitution, while at the same time participating in the disruption of crypt architecture and generating bystander tissue damage, roles that impede and promote the development of the IBD, respectively.
Under the influence of neutrophils, other phagocytic cells such as monocytes and macrophages remove cell debris and help to distinguish damaged areas from tissue areas less affected by acute inflammation [76].
2.1.3. Macrophages in the Gut in Steady State Conditions
Macrophages are highly plastic cells, and their functions depend on their developmental ontogeny and surrounding environment [77,78,79][77][78][79] (Figure 2). During embryonic development, self-maintaining tissue-resident macrophages derive from the yolk sac and fetal liver progenitors [80,81,82,83][80][81][82][83]. After birth, blood circulating bone marrow-derived monocytes are recruited to tissues, where they differentiate to macrophages, replenishing tissue-resident populations and adopting a phenotype conditioned by the local tissue environment [82,84,85,86][82][84][85][86].
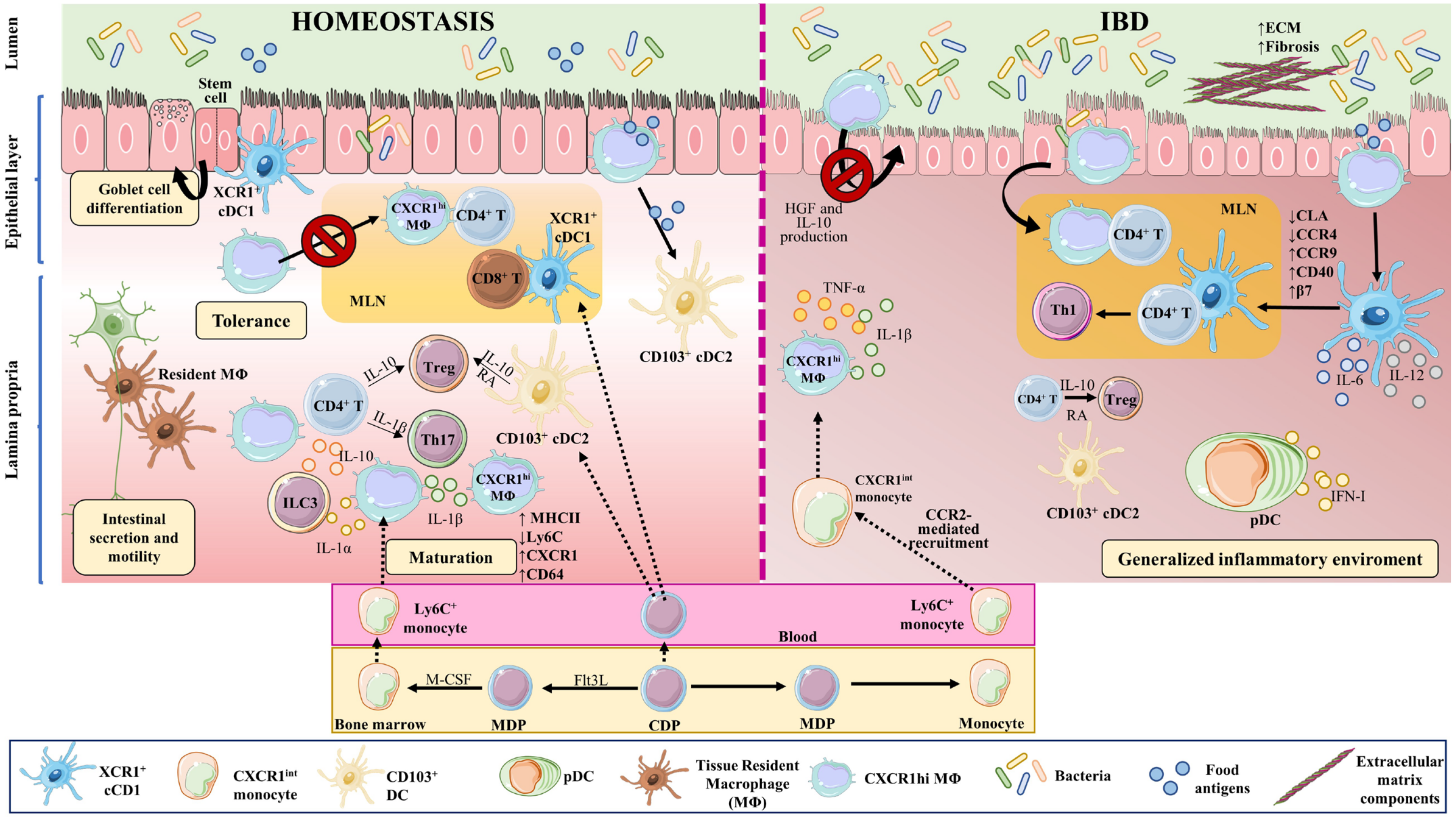
Figure 2. Macrophages and dendritic cell in homeostasis and IBD. Macrophages and DCs play important roles in homeostasis and in the development of IBD by phagocytosing cellular debris, producing cytokines, regulating tissue repair, and interacting with other cells. In the intestine, monocyte-derived macrophages (MΦ) are more abundant than tissue-resident macrophages of embryonic origin. Both perform phagocytosis, produce cytokines, and interact with other cells. Upon weaning, bone marrow-derived monocytes egress from the circulation and extravasate into the tissue, where they undergo differentiation and maturation (downregulation of Ly6C, production of MHCII, and increased expression of CX3CR1). In homeostasis, tissue-resident macrophages in the muscularis externa interact with enteric and myenteric neurons controlling intestinal secretion and motility, while in the lamina propria, macrophages provide signals to intestinal stem cells that give rise to goblet cells, Paneth cells, and intestinal epithelial cells. These macrophages also modulate T cell activities and functions, via the secretion of IL-10 for Tregs and IL-1β for Th17 cells. In addition, they affect ILC3 cells through the production of IL-1α and IL-1β. The migration of antigen-loaded CX3CR1high intestinal macrophages to mesenteric lymph nodes is impaired by intestinal microbiota, thus affecting antigen presentation to T cells and effectively sustaining tolerance towards commensal bacteria. On the other hand, XCR1+ DCs play a tolerogenic role upon recognizing commensal bacterial components, while CD103+ cDC2s seem to be important for initiating oral tolerance through their capacity to generate RA and IL-10. In IBD, large numbers of Ly6Chigh inflammatory monocytes are recruited to the intestine in a CCR2-dependent manner, becoming pro-inflammatory effector cells. These inflammatory macrophages produce TNFα, IL-6, and iNOS, and directly cause the onset and development of fibrosis through a disproportionate accumulation of ECM. The intestinal microbiota is impaired during chronic colitis, and CX3CR1high macrophages can change their habits and migrate to lymph nodes. CD103+ cDC numbers are significantly reduced in the inflamed and uninflamed intestine in IBD; however, activated DCs can release inflammatory cytokines, in addition to type I IFN produced by pDCs. All these phenomena contribute to a generalized inflammation. Part of the figure was generated by using pictures from Servier Medical Art.
Both tissue-resident and monocyte-derived macrophages perform the typical macrophage functions of phagocytosis, cytokine production, and host interaction [87]. In the intestine, monocyte-derived macrophages are more abundant than macrophages of embryonic origin [77]; however, distinct subsets of embryonic-derived macrophages remain in the intestine even after weaning [88].
Upon weaning, bone marrow-derived monocytes egress from the circulation and extravasate into the tissue, contributing to macrophage replenishment in the intestine [89]. After extravasation, monocytes undergo differentiation and maturation processes, acquiring a more macrophage-like phenotype. These changes include downregulation of Ly6C generation, production of class II major histocompatibility complex molecules (MHCII), and increased expression of C-X3-C motif chemokine receptor1 (CX3CR1); monocyte-to-macrophage transition thus involves a switch from a Ly6Chigh, CX3CR1int state to a mature Ly6C−, CX3CR1high, CD64+, MHCII+ state [77,90,91][77][90][91].
In steady-state conditions, intestinal macrophages are distributed throughout the gut structure, including the lamina propria, submucosa and the muscularis externa [92].
Distal to the gut lumen, in the muscularis externa, are located long-lived, bipolar, and stellate self-renewing embryonic-derived macrophages [93]. Muscularis macrophages interact with enteric and myenteric neurons, influencing enteric neurons [94], and controlling intestinal functions including secretion and motility [95,96,97][95][96][97].
The macrophages of the lamina propria are rounded monocyte-derived cells that are constantly replenished [89,94][89][94]. These macrophages are short lived and are distributed close to the lumen, where they constantly encounter the gut microbiota [93,98][93][98] and contribute to oral tolerance [99,100][99][100]. In this layer, macrophages provide signals to the intestinal stem cells, which give rise to goblet cells, Paneth cells, and intestinal epithelial cells [101,102][101][102]. These macrophages also modulate regulatory T cell (Treg) activity and function via the secretion of IL-10 [103] and T helper (Th)17 cells by providing IL-1β [104]. Lamina propria macrophages thus support intestinal homeostasis through a mix of phagocytic and antibacterial functions, immune modulation, and tissue repair [105,106][105][106].
Intestinal-tissue macrophage phenotypes and functions depend on the microbiota and their metabolites and on microenvironmental cues [77,107,108,109][77][107][108][109]. Intestinal macrophages can thus be classified into three distinct populations according to their homeostatic function: host defense, wound healing, and immune regulation [110].
Host defense macrophages have microbicidal activity and, upon stimulation with interferon (IFN)γ or TNF-α, generate cytokines that are commonly secreted by T cells, natural killer cells, or antigen presenting cells (APC). Wound healing macrophages develop upon contact with IL-4 released by T cells or granulocytes, and play a role in tissue repair. Regulatory macrophages, which have an anti-inflammatory function, are generated in response to several stimuli, including IL-10, glucocorticoids, and apoptotic cells [110].
Intestinal macrophages need to combat invading pathogenic bacteria while tolerating beneficial probiotic bacteria [111]. To discern between commensal and harmful bacteria, macrophages recognize harmful bacteria through PRRs such as TLRs and NOD-like receptors (NLRs) [110,112,113][110][112][113]. Intestinal macrophages are excellent phagocytes of harmful bacteria, but upon engulfing or recognizing harmful bacteria they produce low amounts of pro-inflammatory cytokines [114,115,116,117][114][115][116][117]. In contrast, they naturally produce elevated amounts of IL-10, which is associated with their reduced response to TLR-triggering [103,114,118,119,120[103][114][118][119][120][121][122],121,122], suggesting a somewhat anti-inflammatory phenotype for resident intestinal macrophages, which avoid bacteria-activated inflammation in the gut under steady state conditions [117,123][117][123].
In the gut, IL-34 and CSF-1 (also called M-CSF)—both ligands for the CSF-1 receptor (CSF-1R)—promote monocyte and macrophage differentiation [124[124][125],125], and mice lacking CSF-1 or CSF-1R are deficient for tissue macrophages [126,127,128,129][126][127][128][129]. Supporting this, administration of anti-CSFR antibodies reduces macrophage numbers [130], while recombinant CSF-1 increases intestinal macrophage infiltration [131].
Intestinal macrophages are involved in phagocytosis and the clearance of apoptotic cells [132[132][133],133], including apoptotic IECs, helping to maintain epithelial barrier integrity under steady state conditions [134].
CX3CR1high intestinal macrophages sense and take up bacterial antigens from the intestinal lumen through their transepithelial dendrites [135,136,137,138,139,140][135][136][137][138][139][140]. In homeostasis, intestinal microbiota inhibit the migration of antigen-loaded CX3CR1high intestinal macrophages to mesenteric lymph nodes, thereby also inhibiting antigen presentation to T cells and effectively sustaining tolerance towards commensal bacteria. When the intestinal microbiota is disturbed or under chronic colitis conditions, CX3CR1high macrophages can change their habits and migrate to lymph nodes [121].
Intestinal macrophages also regulate other immune cells. CX3CR1high macrophages capture soluble food antigens and transfer them to CD103+ dendritic cells, promoting antigen presentation and food tolerance [141,142][141][142]. Lamina propria macrophages produce IL-10, which promotes the differentiation of Forkhead Box P3 (Foxp3)+ Tregs [103[103][118][143],118,143], and also produce IL-1β, which acts on Th17 cells [104]. Intestinal macrophages also produce IL-1α and IL-1β in response to commensal microbiota, affecting Group 3 innate lymphoid cells (ILC3), and GM-CSF, which acts on macrophages and dendritic cells to maintain Treg homeostasis [107]. CX3CR1+ mononuclear phagocytes prime T cells and promote Th17 cell differentiation [144]. Taken together, these observations show that intestinal macrophages and their secreted cytokines regulate T cell responses in the gut.
2.1.4. Macrophages in the Pathogenesis of IBD
In IBD, the local release of PAMPs and DAMPs at the site of injury triggers intestinal inflammation. During this process, large numbers of Ly6Chigh inflammatory monocytes are recruited to the intestinal tissue in a process dependent on C-C motif chemokine ligand 2 (CCR2) [89[89][90][145][146],90,145,146], which is also known as monocyte chemoattractant protein (MCP)-1. Lack of CCR2 in mice abrogates the recruitment of TLR2+ CCR2+ Gr-1+, TNF-α-producing macrophages to the inflamed intestine [147], and reduces symptoms of DSS-induced colitis [147,148][147][148] (Figure 2).
Monocyte migration to the lamina propria is also controlled by IL-8 and transforming growth factor (TGF)-β, constitutively generated by mucosal epithelial cells [149].
Mouse models of IBD reveal significantly elevated numbers of macrophages in the colon, characterized by an increase in the proportion of Ly6C+ macrophages relative to mature Ly6C− macrophages. This Ly6C+ macrophage recruitment is dependent on the expression of CCR2 [90,147][90][147]. Interestingly, a pronounced elevation in colonic macrophage numbers, and an increased proportion of Ly6C+MHCII+ monocyte-derived macrophages are key features of spontaneous colitis in IL-10R-deficient mice on the C57BL/6 background [150].
IBD patients also have an increased number of pro-inflammatory macrophages [90[90][151][152],151,152], and pediatric IBD patients show accumulation of activated mucosal macrophages [153]. Usually, these macrophages have augmented expression of pro-inflammatory molecules such as TNF-α, IL-1β, IL-6, and inducible nitric oxide synthase (iNOS) [151,154][151][154]. Recruited Ly6Chigh monocytes in IBD upregulate TLR2 and NOD2, which increases their sensitivity to bacteria and triggers their differentiation to pro-inflammatory effector cells [122]. These inflammatory macrophages produce TNF-α, IL-6, and iNOS [110,155][110][155]. Resident CX3CR1high macrophages maintain their anti-inflammatory phenotype even when sharing the intestine with Ly6Chigh inflammatory macrophages [90,116][90][116].
In steady state conditions, CX3CR1high resident macrophages, which are highly phagocytic and MHCIIhigh but resilient to TLR-stimulation and constitutively IL-10 producers, are accompanied by a small population of CX3CR1int cells, mainly resulting in a CX3CR1high resident macrophage population. CX3CR1int cells give rise to CX3CR1high macrophage. In IBD, this CX3CR1int to CX3CR1high macrophage conversion is diminished, leading to the accumulation of TLR-reactive inflammatory CX3CR1int macrophages [90].
CX3CR1 and its ligand CX3CL1 are upregulated in the colon of IBD mice and seem to play an important role in the disease [156], with CX3CR1 and CX3CL1 polymorphisms in patients linked to the clinical manifestations of IBD [157,158][157][158]. However, it is unclear if they play a protective or harmful role, since their deficiency protects from [138] or aggravates [156] experimentally induced colitis depending on the study.
Macrophage-expressed IL-10 and its receptor IL-10R support intestinal homeostasis and are implicated in the development of IBD [159,160,161,162,163,164,165][159][160][161][162][163][164][165]. IL-10–IL-10R-signaling mediates the differentiation and function of intestinal macrophages in mice and IBD patients [166]. The absence of IL-10 in mice provokes a shift from the resident CX3CR1high macrophage phenotype in the colon to a pro-inflammatory phenotype [121]. Interestingly, specific depletion of IL-10 in CX3CR1high intestinal macrophages has no effect on intestinal homeostasis or Treg regulation [121], but the intestinal macrophage-specific lack of IL-10R alters intestinal homeostasis and provokes severe gut inflammation [121,167][121][167]. The absence of Il-10ra in intestinal macrophages promotes the production of IL-23, which in turn mediates IL-22 secretion by Th17 and ILC3 cells. IL-22 activates IECs to express an antimicrobial peptide that induces neutrophil recruitment, promoting IBD [167]. Moreover, colitis symptoms are ameliorated by manipulation of the microbiota to increase IL-10 producing macrophage numbers [109].
Lamina propria macrophages produce the chemokine CCL8, recruiting circulating Ly6Chigh monocytes in IBD [122,168][122][168].
Intestinal macrophages are also important for epithelial tissue repair. These macrophages produce molecules that control epithelial regeneration [169]. As mentioned, intestinal macrophages in the pericryptal stem cell niche stimulate neighboring colonic epithelial progenitors and promote epithelial recuperation after injury [170]. Epithelial recovery is also stimulated by intestinal macrophage-derived IL-10, through the stimulation of the CREB/WISP-1 pathway in epithelial cells [171,172][171][172] or by macrophage production of hepatic growth factor (HGF). Curiously, CD-patient-derived macrophages show diminished HGF secretion, possibly affecting epithelial restoration in these patients [173]. Classically, two main polarized macrophage phenotypes have been proposed, pro-inflammatory M1 and anti-inflammatory M2 [174]. Tissue repairing M2-like macrophages promote stimulation of wingless-related integration site (WNT) signaling in response to differentiation driven by signal transducer and activator of transcription (STAT)-6 [175] through a mechanism dependent on the alarmin IL-33 [176]. STAT6-dependent M2-like macrophage differentiation promotes the stimulation of WNT signaling to promote tissue repair [175]. The alarmin IL-33 also favors protective M2-like macrophages polarization and subsequent mucosal repair [176,177][176][177]. Another study showed that altered α4β7-mediated intestinal-homing of non-classical monocytes might reduce the number of wound-healing macrophages, leading to impaired intestinal wound healing [178].
Upon tissue injury, macrophages are activated to combat microbiota invasion through phagocytosis and to facilitate repair of the damaged tissue. However, when macrophages are improperly activated, they directly cause the onset and development of fibrosis [179,180][179][180]. Fibrosis is a disproportionate accumulation of extracellular matrix (ECM) components such as collagen [181]. Excessive fibrosis produces a non-optimal tissue architecture, and intestinal fibrosis is a common problematic characteristic of IBD [182,183][182][183]. Advanced intestinal fibrosis frequently results in intestinal strictures [106]. Macrophages promote myofibroblast-mediated fibrosis by producing TGF-β1, connective tissue growth factor (CTGF), and fibroblast activation protein (FAP). They also undergo macrophage-to-myofibroblast transition (MMT), favoring myofibroblast accumulation and excess ECM production [106]. In contrast, intestinal macrophages can restrict intestinal fibrosis by promoting myofibroblast senescence, degrading the ECM, and clearing profibrotic components [184]. Macrophages are not the only source of gut fibrosis in IBD; IL-34, which is overproduced in IBD and mediates macrophage maturation [125], also activates collagen synthesis by gut fibroblasts [185].
In summary, macrophages play an important role in homeostasis and in the development of IBD [121[121][150][166][167],150,166,167], both in mouse models and in patients, by phagocytosing cellular debris, producing multiple cytokines, and regulating tissue repair [105].
2.1.5. Innate Lymphoid Cells in IBD
Another important component of the innate immune system in the intestine is the population of innate lymphoid cells (ILCs). ILCs are important mediators of antimicrobial defense and contribute to organ development, tissue protection and regeneration, and mucosal homeostasis [186,187][186][187]. These cells belong to the innate immune system, but are derived from the same common lymphoid progenitor population as lymphocytes [188,189][188][189]. ILCs act early in the immune response by replying quickly to cytokines and other signals produced by other cells [188,190,191,192,193,194][188][190][191][192][193][194]. Recent discoveries have highlighted the essential role of ILCs in intestinal mucosal homeostasis and IBD [195,196,197,198,199,200][195][196][197][198][199][200]. However, they will be not discussed in this reviewere since weresearchers and others have recently reviewed their role in IBD [194,201,202,203,204][194][201][202][203][204].
2.1.6. Dendritic Cells in Homeostasis
DCs link the innate and adaptive immune systems by presenting antigens to and activating T cells. Several DC subtypes are derived from a specific common dendritic progenitor (CDP). CDPs generate plasmacytoid DCs (pDCs) in bone marrow, as well as pre-DCs that circulate in the blood and give rise to conventional or classical DCs (cDCs) in lymphoid and nonlymphoid organs [205,206,207,208,209][205][206][207][208][209]. The production of pDCs versus cDCs is determined by the growth factor fms-like tyrosine kinase 3 ligand (Flt3L) [210] (Figure 2).
Monocytes originate in the bone marrow and circulate in blood. DCs can also originate from circulating monocytes after monocyte migration to inflamed tissues, where they differentiate to macrophages or monocyte-derived DCs through the action of M-CSF or GM-CSF, respectively. Monocyte-derived DCs belong to the mononuclear phagocyte system (MPS), and are better APCs than monocytes [211,212][211][212]. Through their function as professional APCs and their capacity to release cytokines, DCs play important roles in initiating immune responses to invading pathogens; cDCs and monocyte-derived DCs are powerful APCs, whereas pDCs specialize in secreting type I IFN [213]. Moreover, cDC subpopulations included chemokine (C motif) receptor 1 (XCR1)+ cDC1s and signal-regulatory protein alpha (SIPRα)+ cDC2s [205,206,207,208,209][205][206][207][208][209]. cDC2s can be subclassified as CD103+ or CD103−. These DC subsets possess distinctive functional, phenotypical, and transcriptional features. While cDC1 are excellent APCs to cytotoxic T cells, cDC2s are more similar to pDCs in their capacity to polarize CD4+ T cell responses and to promote anti-viral responses via type I IFN [12].
Under physiological conditions, immature DCs patrol peripheral tissues, where they encounter and take up antigens [214]. Upon activation, maturing DCs increase their expression of MHCII and the costimulatory molecules CD80, CD86, and CD83 [209,213][209][213] and migrate along a chemokine gradient to draining lymph nodes, where they enter paracortical T cell zones to activate and prime antigen-specific naïve T cells and secrete cytokines [215,216,217,218][215][216][217][218]. In this way, DCs link innate and adaptive immunity by presenting antigens to and activating T cells.
2.1.7. Dendritic Cells in IBD
DCs accumulate in specific gut locations such as Peyer’s patches, isolated lymphoid follicles, and gut-associated lymphoid tissues. Like macrophages, DCs are constantly replenished from bone marrow-derived progenitors [88] (Figure 2).
Like some macrophages, DCs take up soluble food antigens directly from the intestinal lumen [136], but also take up food antigens from epithelial M-cells in the follicle-associated epithelium of Peyer’s patches [219]. As mentioned, DCs also receive antigens from CX3CR1high lamina propria macrophages through gap junctions [141,142][141][142]. In steady state, DCs play a tolerogenic role upon recognizing commensal bacterial components. XCR1+ cDC1s are important for intestinal homeostasis and in particular the expression of XCR1; mice lacking XCR1 in cDC1 lack intraepithelial and lamina propria T cell populations, and are more vulnerable to chemically-induced colitis [220].
CD103+ cDC2s seem to be important for initiating oral tolerance, in part through their capacity to generate retinoic acid (RA) required for the development of Foxp3+ Treg cells [221,222][221][222]. Furthermore, mammalian target of rapamycin (mTOR) protein kinase intervenes in the regulation of intestinal homeostasis by enhancing IL-10 production in cDC2s. Indeed, loss of mTOR signaling in DCs blocks IL-10 generation by cDC2s and increases sensitivity to DSS-induced colitis [223].
Colonic DCs display an abnormal immature phenotype in IBD that includes the expression of homing markers [224]. Intestinal DCs from UC patients have diminished expression of cutaneous lymphocyte antigen (CLA) and CCR4, while showing enhanced expression of CCR9 and β7 integrin [225,226][225][226].
DCs from CD-patient mucosa express more CD40 and release more IL-6 and IL-12 than DCs from healthy individuals [224]. In IBD, mucosal DCs show increased expression of TLR2 and TLR4 [225]. CD103+ CD11b+ cDCs are significantly reduced in abundance in the inflamed and uninflamed intestinal tissue of CD patients [227]. pDCs are also found in the inflamed gut, although their specific role is still undetermined [228].
The activation of intestinal CD103+ DCs in IBD patients results in the upregulation of PRRs. Thus, local variations in the gut microbiota may change the balance and regulation signals received by mucosal DCs. Upon activation, DCs are able to release inflammatory cytokines [229]. In summary, DCs play an essential role in IBD pathogenesis.
IBD pathogenesis can be augmented by inappropriate macrophage and DC responses to the microbiota [230]. These responses involve inadequate protection and strengthen pathogenicity.
Intestinal DCs promote tolerance to luminal antigens under physiological conditions, but can develop into an inflammatory response after inflammation or direct stimulation by TLR ligands. In these circumstances, intestinal DCs can release inflammatory cytokines such as IL-12, IL-6, and IL-18 and mediate Th1 responses when triggered. Supporting this, the circulation of IBD patients with active disease contains pDCs that migrate to secondary lymphoid organs, where they produce Th1 cytokines (IL-6, IL-8, and TNF-α), thereby perpetuating disease [230]. In addition, inflamed, and uninflamed intestine of CD patients has a reduced abundance of CD11c+ DCs, conferring an increased capacity to produce Th1/Th2/Th17 responses [231].
The role of cDC2s in IBD is less clear, with some studies pointing to an implication in T cell-mediated colitis [232[232][233],233], while others show no effect of lamina propria CD103+ cDC2s [233,234][233][234]. However, conditional absence of interferon regulatory factor (IRF)4 in mice results in abnormal development of colon lamina propria cDC2s and late initiation of T cell-dependent colitis [232], indicating a role of IRF4-expressing cDC2s in the initial priming of colitogenic T cells. Remarkably, cDC1s may play a protective role in the development of IBD, since lack of these cells increases predisposition to DSS-induced colitis [220,233][220][233].
References
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49.
- Levine, A.; Griffiths, A.; Markowitz, J.; Wilson, D.C.; Turner, D.; Russell, R.K.; Fell, J.; Ruemmele, F.M.; Walters, T.; Sherlock, M.; et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: The Paris classification. Inflamm. Bowel Dis. 2011, 17, 1314–1321.
- Louis, E.; Van Kemseke, C.; Reenaers, C. Necessity of phenotypic classification of inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 2011, 25 (Suppl. 1), S2–S7.
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753.
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42, quiz e30.
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217.
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664.
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359.
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55.
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17.
- Gustafsson, J.K.; Johansson, M.E.V. The role of goblet cells and mucus in intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 785–803.
- Ghilas, S.; O’Keefe, R.; Mielke, L.A.; Raghu, D.; Buchert, M.; Ernst, M. Crosstalk between epithelium, myeloid and innate lymphoid cells during gut homeostasis and disease. Front. Immunol. 2022, 13, 944982.
- Bassler, K.; Schulte-Schrepping, J.; Warnat-Herresthal, S.; Aschenbrenner, A.C.; Schultze, J.L. The Myeloid Cell Compartment-Cell by Cell. Annu. Rev. Immunol. 2019, 37, 269–293.
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293.
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835.
- Mann, E.A.; Saeed, S.A. Gastrointestinal infection as a trigger for inflammatory bowel disease. Curr. Opin. Gastroenterol. 2012, 28, 24–29.
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 2018, 23, 27–40.e7.
- Neurath, M.F.; Leppkes, M. Resolution of ulcerative colitis. Semin. Immunopathol. 2019, 41, 747–756.
- Wen, Z.; Fiocchi, C. Inflammatory bowel disease: Autoimmune or immune-mediated pathogenesis? Clin. Dev. Immunol. 2004, 11, 195–204.
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809.
- Munoz, L.E.; Leppkes, M.; Fuchs, T.A.; Hoffmann, M.; Herrmann, M. Missing in action-The meaning of cell death in tissue damage and inflammation. Immunol. Rev. 2017, 280, 26–40.
- Zhou, G.; Yu, L.; Fang, L.; Yang, W.; Yu, T.; Miao, Y.; Chen, M.; Wu, K.; Chen, F.; Cong, Y.; et al. CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 2018, 67, 1052–1063.
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141.
- Mateer, S.W.; Mathe, A.; Bruce, J.; Liu, G.; Maltby, S.; Fricker, M.; Goggins, B.J.; Tay, H.L.; Marks, E.; Burns, G.; et al. IL-6 Drives Neutrophil-Mediated Pulmonary Inflammation Associated with Bacteremia in Murine Models of Colitis. Am. J. Pathol. 2018, 188, 1625–1639.
- Salem, M.; El Azreq, M.A.; Pelletier, J.; Robaye, B.; Aoudjit, F.; Sevigny, J. Exacerbated intestinal inflammation in P2Y6 deficient mice is associated with Th17 activation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2595–2605.
- Kamp, M.E.; Shim, R.; Nicholls, A.J.; Oliveira, A.C.; Mason, L.J.; Binge, L.; Mackay, C.R.; Wong, C.H. G Protein-Coupled Receptor 43 Modulates Neutrophil Recruitment during Acute Inflammation. PLoS ONE 2016, 11, e0163750.
- Correa, R.O.; Vieira, A.; Sernaglia, E.M.; Lancellotti, M.; Vieira, A.T.; Avila-Campos, M.J.; Rodrigues, H.G.; Vinolo, M.A.R. Bacterial short-chain fatty acid metabolites modulate the inflammatory response against infectious bacteria. Cell. Microbiol. 2017, 19, e12720.
- Bedouhene, S.; Liu, M.; Senani, N.; Boussetta, T.; Pintard, C.; Dang, P.M.; El-Benna, J. Prolyl-Isomerase Pin1 Controls Key fMLP-Induced Neutrophil Functions. Biomedicines 2021, 9, 1130.
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137.
- Matoszka, N.; Dzialo, J.; Tokarz-Deptula, B.; Deptula, W. NET and NETosis—New phenomenon in immunology. Postepy. Hig. Med. Dosw. 2012, 66, 437–445.
- Dinallo, V.; Marafini, I.; Di Fusco, D.; Laudisi, F.; Franze, E.; Di Grazia, A.; Figliuzzi, M.M.; Caprioli, F.; Stolfi, C.; Monteleone, I.; et al. Neutrophil Extracellular Traps Sustain Inflammatory Signals in Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 772–784.
- Kaur, A.; Goggolidou, P. Ulcerative colitis: Understanding its cellular pathology could provide insights into novel therapies. J. Inflamm. 2020, 17, 15.
- Scannell, M.; Flanagan, M.B.; de Stefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605.
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628.
- McCracken, J.M.; Allen, L.A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, 15–23.
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639.
- Chen, F.; Liu, Y.; Shi, Y.; Zhang, J.; Liu, X.; Liu, Z.; Lv, J.; Leng, Y. The emerging role of neutrophilic extracellular traps in intestinal disease. Gut Pathog. 2022, 14, 27.
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287.
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.T. Neutrophil Extracellular Traps in Inflammatory Bowel Disease: Pathogenic Mechanisms and Clinical Translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333.
- Bennike, T.B.; Carlsen, T.G.; Ellingsen, T.; Bonderup, O.K.; Glerup, H.; Bogsted, M.; Christiansen, G.; Birkelund, S.; Stensballe, A.; Andersen, V. Neutrophil Extracellular Traps in Ulcerative Colitis: A Proteome Analysis of Intestinal Biopsies. Inflamm. Bowel Dis. 2015, 21, 2052–2067.
- Maronek, M.; Gromova, B.; Liptak, R.; Konecna, B.; Pastorek, M.; Cechova, B.; Harsanyova, M.; Budis, J.; Smolak, D.; Radvanszky, J.; et al. Extracellular DNA Correlates with Intestinal Inflammation in Chemically Induced Colitis in Mice. Cells 2021, 10, 81.
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal Myeloperoxidase as a Biomarker for Inflammatory Bowel Disease. Cureus 2017, 9, e1004.
- Abd El Hafez, A.; Mohamed, A.S.; Shehta, A.; Sheta, H. Neutrophil extracellular traps-associated protein peptidyl arginine deaminase 4 immunohistochemical expression in ulcerative colitis and its association with the prognostic predictors. Pathol. Res. Pract. 2020, 216, 153102.
- Kaluzna, A.; Olczyk, P.; Komosinska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400.
- Zhang, T.; Mei, Y.; Dong, W.; Wang, J.; Huang, F.; Wu, J. Evaluation of protein arginine deiminase-4 inhibitor in TNBS- induced colitis in mice. Int. Immunopharmacol. 2020, 84, 106583.
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohn’s Colitis 2020, 14, 240–253.
- Lin, E.Y.; Lai, H.J.; Cheng, Y.K.; Leong, K.Q.; Cheng, L.C.; Chou, Y.C.; Peng, Y.C.; Hsu, Y.H.; Chiang, H.S. Neutrophil Extracellular Traps Impair Intestinal Barrier Function during Experimental Colitis. Biomedicines 2020, 8, 275.
- Dong, W.; Liu, D.; Zhang, T.; You, Q.; Huang, F.; Wu, J. Oral delivery of staphylococcal nuclease ameliorates DSS induced ulcerative colitis in mice via degrading intestinal neutrophil extracellular traps. Ecotoxicol. Environ. Saf. 2021, 215, 112161.
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257.
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747.
- Wera, O.; Lancellotti, P.; Oury, C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases. J. Clin. Med. 2016, 5, 118.
- Zhou, G.X.; Liu, Z.J. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J. Dig. Dis. 2017, 18, 495–503.
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby-Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820.
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and Inflammasome Inhibition for the Treatment of Inflammatory Skin Diseases. Front. Pharmacol. 2017, 8, 278.
- Mao, L.; Kitani, A.; Strober, W.; Fuss, I.J. The Role of NLRP3 and IL-1beta in the Pathogenesis of Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2566.
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757.
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913.
- Laan, M.; Prause, O.; Miyamoto, M.; Sjostrand, M.; Hytonen, A.M.; Kaneko, T.; Lotvall, J.; Linden, A. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur. Respir. J. 2003, 21, 387–393.
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267.
- Stark, M.A.; Huo, Y.; Burcin, T.L.; Morris, M.A.; Olson, T.S.; Ley, K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 2005, 22, 285–294.
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends. Immunol. 2019, 40, 635–647.
- Danne, C.; Michaudel, C.; Skerniskyte, J.; Planchais, J.; Magniez, A.; Agus, A.; Michel, M.L.; Lamas, B.; Da Costa, G.; Spatz, M.; et al. CARD9 in neutrophils protects from colitis and controls mitochondrial metabolism and cell survival. Gut 2022.
- Kang, L.; Fang, X.; Song, Y.H.; He, Z.X.; Wang, Z.J.; Wang, S.L.; Li, Z.S.; Bai, Y. Neutrophil-Epithelial Crosstalk During Intestinal Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1257–1267.
- McCourt, M.; Wang, J.H.; Sookhai, S.; Redmond, H.P. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch. Surg. 1999, 134, 1325–1331, discussion 1331–1332.
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874.
- Seo, D.H.; Che, X.; Kim, S.; Kim, D.H.; Ma, H.W.; Kim, J.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; Cheon, J.H. Triggering Receptor Expressed on Myeloid Cells-1 Agonist Regulates Intestinal Inflammation via Cd177(+) Neutrophils. Front. Immunol. 2021, 12, 650864.
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950.
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 2000, 68, 9–14.
- Cunliffe, R.N.; Kamal, M.; Rose, F.R.; James, P.D.; Mahida, Y.R. Expression of antimicrobial neutrophil defensins in epithelial cells of active inflammatory bowel disease mucosa. J. Clin. Pathol. 2002, 55, 298–304.
- Yui, S.; Nakatani, Y.; Mikami, M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol. Pharm. Bull. 2003, 26, 753–760.
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212.
- Ozaki, R.; Kobayashi, T.; Okabayashi, S.; Nakano, M.; Morinaga, S.; Hara, A.; Ohbu, M.; Matsuoka, K.; Toyonaga, T.; Saito, E.; et al. Histological Risk Factors to Predict Clinical Relapse in Ulcerative Colitis with Endoscopically Normal Mucosa. J. Crohn’s Colitis 2018, 12, 1288–1294.
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763.
- Shinoda, M.; Shin-Ya, M.; Naito, Y.; Kishida, T.; Ito, R.; Suzuki, N.; Yasuda, H.; Sakagami, J.; Imanishi, J.; Kataoka, K.; et al. Early-stage blocking of Notch signaling inhibits the depletion of goblet cells in dextran sodium sulfate-induced colitis in mice. J. Gastroenterol. 2010, 45, 608–617.
- Leiper, K.; Campbell, B.J.; Jenkinson, M.D.; Milton, J.; Yu, L.G.; Democratis, J.; Rhodes, J.M. Interaction between bacterial peptides, neutrophils and goblet cells: A possible mechanism for neutrophil recruitment and goblet cell depletion in colitis. Clin. Sci. 2001, 101, 395–402.
- Bilyy, R.; Fedorov, V.; Vovk, V.; Leppkes, M.; Dumych, T.; Chopyak, V.; Schett, G.; Herrmann, M. Neutrophil Extracellular Traps Form a Barrier between Necrotic and Viable Areas in Acute Abdominal Inflammation. Front. Immunol. 2016, 7, 424.
- Bain, C.C.; Schridde, A. Origin, Differentiation, and Function of Intestinal Macrophages. Front. Immunol. 2018, 9, 2733.
- Bleriot, C.; Chakarov, S.; Ginhoux, F. Determinants of Resident Tissue Macrophage Identity and Function. Immunity 2020, 52, 957–970.
- Lis-Lopez, L.; Bauset, C.; Seco-Cervera, M.; Cosin-Roger, J. Is the Macrophage Phenotype Determinant for Fibrosis Development? Biomedicines 2021, 9, 1747.
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551.
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460.
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35.
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440.
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147.
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8.
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449.
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415.e13.
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937.
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510.
- Tamoutounour, S.; Henri, S.; Lelouard, H.; de Bovis, B.; de Haar, C.; van der Woude, C.J.; Woltman, A.M.; Reyal, Y.; Bonnet, D.; Sichien, D.; et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012, 42, 3150–3166.
- Delfini, M.; Stakenborg, N.; Viola, M.F.; Boeckxstaens, G. Macrophages in the gut: Masters in multitasking. Immunity 2022, 55, 1530–1548.
- Viola, M.F.; Boeckxstaens, G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut 2021, 70, 1383–1395.
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391.
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543.
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.J.L.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 2018, 215, 1507–1518.
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 2014, 158, 300–313.
- Hume, D.A.; Perry, V.H.; Gordon, S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localisation of antigen F4/80: Macrophages associated with epithelia. Anat. Rec. 1984, 210, 503–512.
- Hayashi, A.; Sato, T.; Kamada, N.; Mikami, Y.; Matsuoka, K.; Hisamatsu, T.; Hibi, T.; Roers, A.; Yagita, H.; Ohteki, T.; et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe 2013, 13, 711–722.
- Danne, C.; Ryzhakov, G.; Martinez-Lopez, M.; Ilott, N.E.; Franchini, F.; Cuskin, F.; Lowe, E.C.; Bullers, S.J.; Arthur, J.S.C.; Powrie, F. A Large Polysaccharide Produced by Helicobacter hepaticus Induces an Anti-inflammatory Gene Signature in Macrophages. Cell Host Microbe 2017, 22, 733–745.e5.
- Hiemstra, I.H.; Beijer, M.R.; Veninga, H.; Vrijland, K.; Borg, E.G.; Olivier, B.J.; Mebius, R.E.; Kraal, G.; den Haan, J.M. The identification and developmental requirements of colonic CD169(+) macrophages. Immunology 2014, 142, 269–278.
- Sehgal, A.; Donaldson, D.S.; Pridans, C.; Sauter, K.A.; Hume, D.A.; Mabbott, N.A. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun. 2018, 9, 1272.
- Hadis, U.; Wahl, B.; Schulz, O.; Hardtke-Wolenski, M.; Schippers, A.; Wagner, N.; Muller, W.; Sparwasser, T.; Forster, R.; Pabst, O. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011, 34, 237–246.
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258.
- He, X.; Tan, S.; Shao, Z.; Wang, X. Latitudinal and longitudinal regulation of tissue macrophages in inflammatory diseases. Genes Dis. 2022, 9, 1194–1207.
- Yao, H.; Tang, G. Macrophages in intestinal fibrosis and regression. Cell Immunol. 2022, 381, 104614.
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288.
- Spindler, M.P.; Siu, S.; Mogno, I.; Li, Z.; Yang, C.; Mehandru, S.; Britton, G.J.; Faith, J.J. Human gut microbiota stimulate defined innate immune responses that vary from phylum to strain. Cell Host Microbe 2022, 30, 1481–1498.e5.
- Zegarra Ruiz, D.F.; Kim, D.V.; Norwood, K.; Saldana-Morales, F.B.; Kim, M.; Ng, C.; Callaghan, R.; Uddin, M.; Chang, L.C.; Longman, R.S.; et al. Microbiota manipulation to increase macrophage IL-10 improves colitis and limits colitis-associated colorectal cancer. Gut Microbes 2022, 14, 2119054.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969.
- Ruder, B.; Becker, C. At the Forefront of the Mucosal Barrier: The Role of Macrophages in the Intestine. Cells 2020, 9, 2162.
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080.
- Bortoluci, K.R.; Medzhitov, R. Control of infection by pyroptosis and autophagy: Role of TLR and NLR. Cell Mol. Life Sci. 2010, 67, 1643–1651.
- Rivollier, A.; He, J.; Kole, A.; Valatas, V.; Kelsall, B.L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 2012, 209, 139–155.
- Smith, P.D.; Smythies, L.E.; Mosteller-Barnum, M.; Sibley, D.A.; Russell, M.W.; Merger, M.; Sellers, M.T.; Orenstein, J.M.; Shimada, T.; Graham, M.F.; et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J. Immunol. 2001, 167, 2651–2656.
- Weber, B.; Saurer, L.; Schenk, M.; Dickgreber, N.; Mueller, C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur. J. Immunol. 2011, 41, 773–779.
- Smythies, L.E.; Sellers, M.; Clements, R.H.; Mosteller-Barnum, M.; Meng, G.; Benjamin, W.H.; Orenstein, J.M.; Smith, P.D. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Investig. 2005, 115, 66–75.
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094.
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184.
- Ueda, Y.; Kayama, H.; Jeon, S.G.; Kusu, T.; Isaka, Y.; Rakugi, H.; Yamamoto, M.; Takeda, K. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int. Immunol. 2010, 22, 953–962.
- Zigmond, E.; Bernshtein, B.; Friedlander, G.; Walker, C.R.; Yona, S.; Kim, K.W.; Brenner, O.; Krauthgamer, R.; Varol, C.; Muller, W.; et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 2014, 40, 720–733.
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 2012, 37, 1076–1090.
- Zhou, Z.; Ding, M.; Huang, L.; Gilkeson, G.; Lang, R.; Jiang, W. Toll-like receptor-mediated immune responses in intestinal macrophages; implications for mucosal immunity and autoimmune diseases. Clin. Immunol. 2016, 173, 81–86.
- Lin, W.; Xu, D.; Austin, C.D.; Caplazi, P.; Senger, K.; Sun, Y.; Jeet, S.; Young, J.; Delarosa, D.; Suto, E.; et al. Function of CSF1 and IL34 in Macrophage Homeostasis, Inflammation, and Cancer. Front. Immunol. 2019, 10, 2019.
- Zwicker, S.; Martinez, G.L.; Bosma, M.; Gerling, M.; Clark, R.; Majster, M.; Soderman, J.; Almer, S.; Bostrom, E.A. Interleukin 34: A new modulator of human and experimental inflammatory bowel disease. Clin. Sci. 2015, 129, 281–290.
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120.
- Felix, R.; Cecchini, M.G.; Hofstetter, W.; Elford, P.R.; Stutzer, A.; Fleisch, H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1990, 5, 781–789.
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832.
- Cecchini, M.G.; Dominguez, M.G.; Mocci, S.; Wetterwald, A.; Felix, R.; Fleisch, H.; Chisholm, O.; Hofstetter, W.; Pollard, J.W.; Stanley, E.R. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 1994, 120, 1357–1372.
- MacDonald, K.P.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B.; et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010, 116, 3955–3963.
- Hume, D.A.; Pavli, P.; Donahue, R.E.; Fidler, I.J. The effect of human recombinant macrophage colony-stimulating factor (CSF-1) on the murine mononuclear phagocyte system in vivo. J. Immunol. 1988, 141, 3405–3409.
- Schridde, A.; Bain, C.C.; Mayer, J.U.; Montgomery, J.; Pollet, E.; Denecke, B.; Milling, S.W.F.; Jenkins, S.J.; Dalod, M.; Henri, S.; et al. Tissue-specific differentiation of colonic macrophages requires TGFbeta receptor-mediated signaling. Mucosal Immunol. 2017, 10, 1387–1399.
- Smith, P.D.; Janoff, E.N.; Mosteller-Barnum, M.; Merger, M.; Orenstein, J.M.; Kearney, J.F.; Graham, M.F. Isolation and purification of CD14-negative mucosal macrophages from normal human small intestine. J. Immunol. Methods 1997, 202, 1–11.
- Cummings, R.J.; Barbet, G.; Bongers, G.; Hartmann, B.M.; Gettler, K.; Muniz, L.; Furtado, G.C.; Cho, J.; Lira, S.A.; Blander, J.M. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 2016, 539, 565–569.
- Vallon-Eberhard, A.; Landsman, L.; Yogev, N.; Verrier, B.; Jung, S. Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 2006, 176, 2465–2469.
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367.
- Hapfelmeier, S.; Muller, A.J.; Stecher, B.; Kaiser, P.; Barthel, M.; Endt, K.; Eberhard, M.; Robbiani, R.; Jacobi, C.A.; Heikenwalder, M.; et al. Microbe sampling by mucosal dendritic cells is a discrete, MyD88-independent step in DeltainvG S. Typhimurium colitis. J. Exp. Med. 2008, 205, 437–450.
- Medina-Contreras, O.; Geem, D.; Laur, O.; Williams, I.R.; Lira, S.A.; Nusrat, A.; Parkos, C.A.; Denning, T.L. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J. Clin. Investig. 2011, 121, 4787–4795.
- Pabst, O.; Bernhardt, G. The puzzle of intestinal lamina propria dendritic cells and macrophages. Eur. J. Immunol. 2010, 40, 2107–2111.
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258.
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity 2014, 40, 248–261.
- Farache, J.; Koren, I.; Milo, I.; Gurevich, I.; Kim, K.W.; Zigmond, E.; Furtado, G.C.; Lira, S.A.; Shakhar, G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity 2013, 38, 581–595.
- Denning, T.L.; Norris, B.A.; Medina-Contreras, O.; Manicassamy, S.; Geem, D.; Madan, R.; Karp, C.L.; Pulendran, B. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J. Immunol. 2011, 187, 733–747.
- Panea, C.; Farkas, A.M.; Goto, Y.; Abdollahi-Roodsaz, S.; Lee, C.; Koscso, B.; Gowda, K.; Hohl, T.M.; Bogunovic, M.; Ivanov, I.I. Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell. Rep. 2015, 12, 1314–1324.
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317.
- El Sayed, S.; Patik, I.; Redhu, N.S.; Glickman, J.N.; Karagiannis, K.; El Naenaeey, E.S.Y.; Elmowalid, G.A.; Abd El Wahab, A.M.; Snapper, S.B.; Horwitz, B.H. CCR2 promotes monocyte recruitment and intestinal inflammation in mice lacking the interleukin-10 receptor. Sci. Rep. 2022, 12, 452.
- Platt, A.M.; Bain, C.C.; Bordon, Y.; Sester, D.P.; Mowat, A.M. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 2010, 184, 6843–6854.
- Andres, P.G.; Beck, P.L.; Mizoguchi, E.; Mizoguchi, A.; Bhan, A.K.; Dawson, T.; Kuziel, W.A.; Maeda, N.; MacDermott, R.P.; Podolsky, D.K.; et al. Mice with a selective deletion of the CC chemokine receptors 5 or 2 are protected from dextran sodium sulfate-mediated colitis: Lack of CC chemokine receptor 5 expression results in a NK1.1+ lymphocyte-associated Th2-type immune response in the intestine. J. Immunol. 2000, 164, 6303–6312.
- Smythies, L.E.; Maheshwari, A.; Clements, R.; Eckhoff, D.; Novak, L.; Vu, H.L.; Mosteller-Barnum, L.M.; Sellers, M.; Smith, P.D. Mucosal IL-8 and TGF-beta recruit blood monocytes: Evidence for cross-talk between the lamina propria stroma and myeloid cells. J. Leukoc. Biol. 2006, 80, 492–499.
- Redhu, N.S.; Bakthavatchalu, V.; Conaway, E.A.; Shouval, D.S.; Tsou, A.; Goettel, J.A.; Biswas, A.; Wang, C.; Field, M.; Muller, W.; et al. Macrophage dysfunction initiates colitis during weaning of infant mice lacking the interleukin-10 receptor. elife 2017, 6, e27652.
- Rugtveit, J.; Nilsen, E.M.; Bakka, A.; Carlsen, H.; Brandtzaeg, P.; Scott, H. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology 1997, 112, 1493–1505.
- Thiesen, S.; Janciauskiene, S.; Uronen-Hansson, H.; Agace, W.; Hogerkorp, C.M.; Spee, P.; Hakansson, K.; Grip, O. CD14(hi)HLA-DR(dim) macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn’s disease. J. Leukoc. Biol. 2014, 95, 531–541.
- Perminow, G.; Reikvam, D.H.; Lyckander, L.G.; Brandtzaeg, P.; Vatn, M.H.; Carlsen, H.S. Increased number and activation of colonic macrophages in pediatric patients with untreated Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 1368–1378.
- Grimm, M.C.; Pullman, W.E.; Bennett, G.M.; Sullivan, P.J.; Pavli, P.; Doe, W.F. Direct evidence of monocyte recruitment to inflammatory bowel disease mucosa. J. Gastroenterol. Hepatol. 1995, 10, 387–395.
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185.
- Kostadinova, F.I.; Baba, T.; Ishida, Y.; Kondo, T.; Popivanova, B.K.; Mukaida, N. Crucial involvement of the CX3CR1-CX3CL1 axis in dextran sulfate sodium-mediated acute colitis in mice. J. Leukoc. Biol. 2010, 88, 133–143.
- Brand, S.; Hofbauer, K.; Dambacher, J.; Schnitzler, F.; Staudinger, T.; Pfennig, S.; Seiderer, J.; Tillack, C.; Konrad, A.; Goke, B.; et al. Increased expression of the chemokine fractalkine in Crohn’s disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease Phenotype. Am. J. Gastroenterol. 2006, 101, 99–106.
- Sabate, J.M.; Ameziane, N.; Lamoril, J.; Jouet, P.; Farmachidi, J.P.; Soule, J.C.; Harnois, F.; Sobhani, I.; Jian, R.; Deybach, J.C.; et al. The V249I polymorphism of the CX3CR1 gene is associated with fibrostenotic disease behavior in patients with Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2008, 20, 748–755.
- Begue, B.; Verdier, J.; Rieux-Laucat, F.; Goulet, O.; Morali, A.; Canioni, D.; Hugot, J.P.; Daussy, C.; Verkarre, V.; Pigneur, B.; et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am. J. Gastroenterol. 2011, 106, 1544–1555.
- Franke, A.; Balschun, T.; Karlsen, T.H.; Sventoraityte, J.; Nikolaus, S.; Mayr, G.; Domingues, F.S.; Albrecht, M.; Nothnagel, M.; Ellinghaus, D.; et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 2008, 40, 1319–1323.
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045.
- Kotlarz, D.; Beier, R.; Murugan, D.; Diestelhorst, J.; Jensen, O.; Boztug, K.; Pfeifer, D.; Kreipe, H.; Pfister, E.D.; Baumann, U.; et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: Implications for diagnosis and therapy. Gastroenterology 2012, 143, 347–355.
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274.
- Moran, C.J.; Walters, T.D.; Guo, C.H.; Kugathasan, S.; Klein, C.; Turner, D.; Wolters, V.M.; Bandsma, R.H.; Mouzaki, M.; Zachos, M.; et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 115–123.
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J. Exp. Med. 1998, 187, 571–578.
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014, 40, 706–719.
- Bernshtein, B.; Curato, C.; Ioannou, M.; Thaiss, C.A.; Gross-Vered, M.; Kolesnikov, M.; Wang, Q.; David, E.; Chappell-Maor, L.; Harmelin, A.; et al. IL-23-producing IL-10Ralpha-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci. Immunol. 2019, 4, eaau6571.
- Asano, K.; Takahashi, N.; Ushiki, M.; Monya, M.; Aihara, F.; Kuboki, E.; Moriyama, S.; Iida, M.; Kitamura, H.; Qiu, C.H.; et al. Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat. Commun. 2015, 6, 7802.
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67.
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104.
- Morhardt, T.L.; Hayashi, A.; Ochi, T.; Quiros, M.; Kitamoto, S.; Nagao-Kitamoto, H.; Kuffa, P.; Atarashi, K.; Honda, K.; Kao, J.Y.; et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci. Rep. 2019, 9, 1223.
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G.; et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520.
- D’Angelo, F.; Bernasconi, E.; Schafer, M.; Moyat, M.; Michetti, P.; Maillard, M.H.; Velin, D. Macrophages promote epithelial repair through hepatocyte growth factor secretion. Clin. Exp. Immunol. 2013, 174, 60–72.
- Ma, S.; Zhang, J.; Liu, H.; Li, S.; Wang, Q. The Role of Tissue-Resident Macrophages in the Development and Treatment of Inflammatory Bowel Disease. Front. Cell Dev. Biol. 2022, 10, 896591.
- Cosin-Roger, J.; Ortiz-Masia, D.; Calatayud, S.; Hernandez, C.; Esplugues, J.V.; Barrachina, M.D. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2016, 9, 986–998.
- Seo, D.H.; Che, X.; Kwak, M.S.; Kim, S.; Kim, J.H.; Ma, H.W.; Kim, D.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci. Rep. 2017, 7, 851.
- Waddell, A.; Vallance, J.E.; Moore, P.D.; Hummel, A.T.; Wu, D.; Shanmukhappa, S.K.; Fei, L.; Washington, M.K.; Minar, P.; Coburn, L.A.; et al. IL-33 Signaling Protects from Murine Oxazolone Colitis by Supporting Intestinal Epithelial Function. Inflamm. Bowel Dis. 2015, 21, 2737–2746.
- Schleier, L.; Wiendl, M.; Heidbreder, K.; Binder, M.T.; Atreya, R.; Rath, T.; Becker, E.; Schulz-Kuhnt, A.; Stahl, A.; Schulze, L.L.; et al. Non-classical monocyte homing to the gut via alpha4beta7 integrin mediates macrophage-dependent intestinal wound healing. Gut 2020, 69, 252–263.
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93.
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257.
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040.
- Lenti, M.V.; Di Sabatino, A. Intestinal fibrosis. Mol. Aspects Med. 2019, 65, 100–109.
- D’Alessio, S.; Ungaro, F.; Noviello, D.; Lovisa, S.; Peyrin-Biroulet, L.; Danese, S. Revisiting fibrosis in inflammatory bowel disease: The gut thickens. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 169–184.
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31.
- Franze, E.; Dinallo, V.; Laudisi, F.; Di Grazia, A.; Di Fusco, D.; Colantoni, A.; Ortenzi, A.; Giuffrida, P.; Di Carlo, S.; Sica, G.S.; et al. Interleukin-34 Stimulates Gut Fibroblasts to Produce Collagen Synthesis. J. Crohn’s Colitis 2020, 14, 1436–1445.
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74.
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725.
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149.
- Ignacio, A.; Breda, C.N.S.; Camara, N.O.S. Innate lymphoid cells in tissue homeostasis and diseases. World. J. Hepatol. 2017, 9, 979–989.
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566.
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301.
- Huang, Y.; Mao, K.; Germain, R.N. Thinking differently about ILCs-Not just tissue resident and not just the same as CD4(+) T-cell effectors. Immunol. Rev. 2018, 286, 160–171.
- Trabanelli, S.; Gomez-Cadena, A.; Salome, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. B Clin. Cytom. 2018, 94, 392–399.
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861.
- Giuffrida, P.; Corazza, G.R.; Di Sabatino, A. Old and New Lymphocyte Players in Inflammatory Bowel Disease. Dig. Dis. Sci. 2018, 63, 277–288.
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2021, 14, 317–330.
- Luo, W.; Tian, L.; Tan, B.; Shen, Z.; Xiao, M.; Wu, S.; Meng, X.; Wu, X.; Wang, X. Update: Innate Lymphoid Cells in Inflammatory Bowel Disease. Dig. Dis. Sci. 2021, 67, 56–66.
- Zhou, W.; Sonnenberg, G.F. Activation and Suppression of Group 3 Innate Lymphoid Cells in the Gut. Trends Immunol. 2020, 41, 721–733.
- Wu, Y.; Shen, J. Innate Lymphoid Cells in Crohn’s Disease. Front. Immunol. 2020, 11, 554880.
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate Lymphoid Cell-Epithelial Cell Modules Sustain Intestinal Homeostasis. Immunity 2020, 52, 452–463.
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618.
- Zeng, B.; Shi, S.; Ashworth, G.; Dong, C.; Liu, J.; Xing, F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019, 10, 315.
- Schulz-Kuhnt, A.; Neurath, M.F.; Wirtz, S.; Atreya, I. Innate Lymphoid Cells as Regulators of Epithelial Integrity: Therapeutic Implications for Inflammatory Bowel Diseases. Front. Med. 2021, 8, 656745.
- Peng, V.; Jaeger, N.; Colonna, M. Innate Lymphoid Cells and Inflammatory Bowel Disease. Adv. Exp. Med. Biol. 2022, 1365, 97–112.
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20.
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656.
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684.
- Dutertre, C.A.; Wang, L.F.; Ginhoux, F. Aligning bona fide dendritic cell populations across species. Cell. Immunol. 2014, 291, 3–10.
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770.
- Gil-Pulido, J.; Zernecke, A. Antigen-presenting dendritic cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 25–31.
- Greter, M.; Helft, J.; Chow, A.; Hashimoto, D.; Mortha, A.; Agudo-Cantero, J.; Bogunovic, M.; Gautier, E.L.; Miller, J.; Leboeuf, M.; et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 2012, 36, 1031–1046.
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489.
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811.
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258.
- Palucka, K.; Banchereau, J. Dendritic cells: A link between innate and adaptive immunity. J. Clin. Immunol. 1999, 19, 12–25.
- Palucka, K.; Banchereau, J. Linking innate and adaptive immunity. Nat. Med. 1999, 5, 868–870.
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684.
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22.
- Lelouard, H.; Fallet, M.; de Bovis, B.; Meresse, S.; Gorvel, J.P. Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 2012, 142, 592–601.e3.
- Ohta, T.; Sugiyama, M.; Hemmi, H.; Yamazaki, C.; Okura, S.; Sasaki, I.; Fukuda, Y.; Orimo, T.; Ishii, K.J.; Hoshino, K.; et al. Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Sci. Rep. 2016, 6, 23505.
- Johansson-Lindbom, B.; Svensson, M.; Pabst, O.; Palmqvist, C.; Marquez, G.; Forster, R.; Agace, W.W. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 2005, 202, 1063–1073.
- Agace, W.W.; Persson, E.K. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 2012, 33, 42–48.
- Ohtani, M.; Hoshii, T.; Fujii, H.; Koyasu, S.; Hirao, A.; Matsuda, S. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 2012, 188, 4736–4740.
- Al-Hassi, H.O.; Mann, E.R.; Sanchez, B.; English, N.R.; Peake, S.T.; Landy, J.; Man, R.; Urdaci, M.; Hart, A.L.; Fernandez-Salazar, L.; et al. Altered human gut dendritic cell properties in ulcerative colitis are reversed by Lactobacillus plantarum extracellular encrypted peptide STp. Mol. Nutr. Food Res. 2014, 58, 1132–1143.
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 2005, 129, 50–65.
- Magnusson, M.K.; Brynjolfsson, S.F.; Dige, A.; Uronen-Hansson, H.; Borjesson, L.G.; Bengtsson, J.L.; Gudjonsson, S.; Ohman, L.; Agnholt, J.; Sjovall, H.; et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. 2016, 9, 171–182.
- Faleiro, R.; Liu, J.; Karunarathne, D.; Edmundson, A.; Winterford, C.; Nguyen, T.H.; Simms, L.A.; Radford-Smith, G.; Wykes, M. Crohn’s disease is facilitated by a disturbance of programmed death-1 ligand 2 on blood dendritic cells. Clin. Transl. Immunology 2019, 8, e01071.
- Sawai, C.M.; Serpas, L.; Neto, A.G.; Jang, G.; Rashidfarrokhi, A.; Kolbeck, R.; Sanjuan, M.A.; Reizis, B.; Sisirak, V. Plasmacytoid Dendritic Cells Are Largely Dispensable for the Pathogenesis of Experimental Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 2475.
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2020, 55, 4–14.
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434.
- Matsuno, H.; Kayama, H.; Nishimura, J.; Sekido, Y.; Osawa, H.; Barman, S.; Ogino, T.; Takahashi, H.; Haraguchi, N.; Hata, T.; et al. CD103+ Dendritic Cell Function Is Altered in the Colons of Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1524–1534.
- Pool, L.; Rivollier, A.; Agace, W.W. Deletion of IRF4 in Dendritic Cells Leads to Delayed Onset of T Cell-Dependent Colitis. J. Immunol. 2020, 204, 1047–1055.
- Muzaki, A.R.; Tetlak, P.; Sheng, J.; Loh, S.C.; Setiagani, Y.A.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-gamma-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016, 9, 336–351.
- Welty, N.E.; Staley, C.; Ghilardi, N.; Sadowsky, M.J.; Igyarto, B.Z.; Kaplan, D.H. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J. Exp. Med. 2013, 210, 2011–2024.
More