Blood pressure is determined by cardiac output and peripheral vascular resistance. The L-type voltage-gated Ca2+ (Cav1.2) channel in small arteries and arterioles plays an essential role in regulating Ca2+ influx, vascular resistance, and blood pressure. Hypertension and preeclampsia are characterized by high blood pressure. In addition, diabetes has a high prevalence of hypertension. The etiology of these disorders remains elusive, involving the complex interplay of environmental and genetic factors. Common to these disorders are oxidative stress and vascular dysfunction. Reactive oxygen species (ROS) derived from NADPH oxidases (NOXs) and mitochondria are primary sources of vascular oxidative stress, whereas dysfunction of the Cav1.2 channel confers increased vascular resistance in hypertension.
- hypertension
- gestational diabetes
- Cav1.2
1. Introduction
2. Cav1.2 in Vascular Smooth Muscle
2.1. Overview of Ca
v
1.2
L-type Ca2+ (Cav) channels are heteromultimeric complexes comprising pore-forming α1c and auxiliary β, α2δ, and γ subunits [27][18]. The α1c subunit possesses four repeat domains (I–IV) linked by intracellular loops and intracellular NH2-/COOH-termini with each domain containing six transmembrane segments (S1–S6) (Figure 1). The ion-conducting pore is formed by S5 and S6 and the loop between them, whereas the voltage sensor is located in the S4 segments [28][19]. The intracellular COOH terminus, along with intracellular loops, plays important roles in Ca2+-dependent inactivation, channel trafficking, phosphorylation and oxidation [28,29,30][19][20][21]. Cav is activated by membrane depolarization and its activation allows Ca2+ influx through the channel pore. The expression of the α1c subunit itself can form functional channels to conduct Ca2+ ions, whereas the incorporation of auxiliary subunits promotes membrane α1c expression and alters biophysical properties of the channel [28,31][19][22]. Cav channels are sensitive to blockades by dihydropyridines (i.e., nifedipine), phenylalkylamines (i.e., verapamil) and benzothiazepines (i.e., diltiazem) [32][23]. There are four types of Cav channels (Cav1.1–1.4). Cav1.2 is predominantly expressed in the heart and in vascular smooth muscle [33,34][24][25]. Ca2+ influx through the channel is a primary trigger of vasoconstriction and also participates in transcriptional regulation. The auxiliary subunits β2, β3, and α2δ1 are expressed in vascular smooth muscle [35,36,37,38][26][27][28][29]. β subunits, lacking membrane-spanning segments, are located intracellularly and interact with the α interaction domain (AID) on the I-II linker of the α1c subunit [39][30]. The α2δ1 subunit is a single gene product bound together by disulfide bonds. Whereas the α2 subunit is extracellular, the δ subunit contains a single transmembrane segment [40][31]. The diversity of Cav1.2 is also conferred by alternative splicing. Smooth muscle contains Cav1.2 exons 1, 8, 9 *, 31/32, and 33 [41,42][32][33].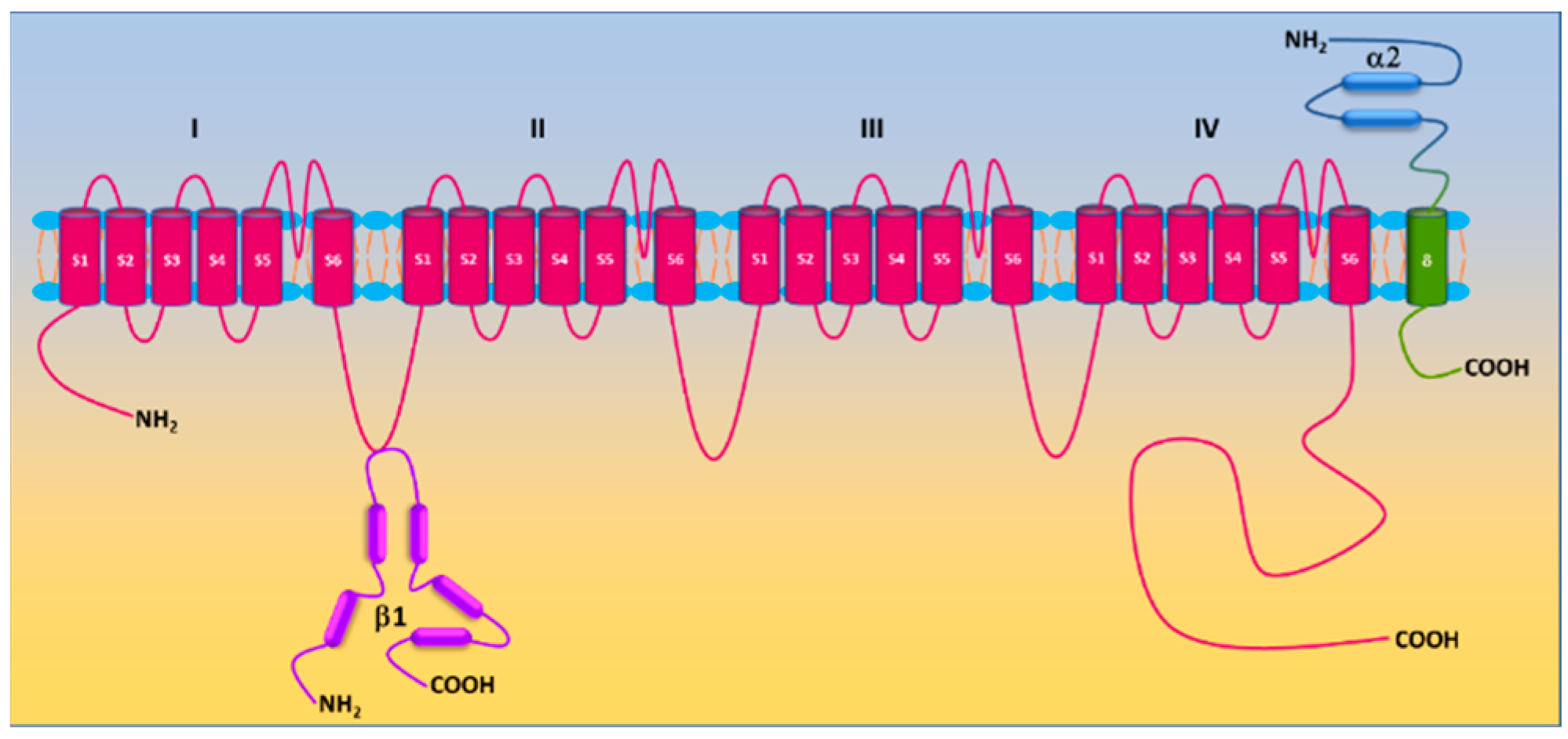
2.2. Regulation of Ca
v
1.2
2.2.1. Regulation by Auxiliary Subunits
The molecular composition of Cav1.2 in vascular smooth muscle cells includes the pore-forming α1c subunit and auxiliary β2/3 and α2/δ1 subunits. The β3 subunit appears to be the principal β isoform in vascular smooth muscle cells [35][26]. Genetic deletion of the β3 subunit resulted in reduced α1c expression in mouse aorta and is associated with a reduction in Ca2+ channel current and a slower inactivation rate [35][26]. This genetic manipulation also attenuates angiotensin II-induced upregulation of Cav1.2 channels in mouse mesenteric arteries and the development of hypertension [38][29].2.2.2. Regulation by Protein Kinases
Protein phosphorylation, a covalent addition of the phosphate group to the side chain of serine, threonine, and tyrosine residues by protein kinases, is a common posttranslational modification to fine tune activities of receptors, ion channels and enzymes. Unsurprisingly, Cav1.2 is a target of protein kinases, and its activity is subject to the regulation by protein phosphorylation. Both α1c and β2 subunits are phosphorylated by protein kinases A (PKA), C (PKC), and G (PKG) [29,46,47,48,49][20][34][35][36][37]. A variety of putative serine/threonine phosphorylation sites have been identified, yet their role in regulating Cav1.2 remains unsettled [50][38]. In vascular smooth muscle cells, the regulation of Cav1.2 by PKA is controversial. PKA is found to either inhibit or enhance Cav1.2 activity [51,52,53,54,55,56,57][39][40][41][42][43][44][45]. The stimulatory effect of PKA on Cav1.2 in vascular smooth muscle cells depends on anchoring adenyl cyclase 5 and PKA by A-kinase anchoring protein 150 (AKAP150) to the proximity of Cav1.2 [56,58][44][46]. Phosphorylation of Ser 1928 in the COOH-terminus of the Cav1.2 α1c subunit is required for PKA-stimulated channel activity in vascular smooth muscle cells [56][44]. Activation of PKG exhibits inhibitory effects on vascular Cav1.2 [52,57,59,60][40][45][47][48]. PKG mediates nitric oxide-induced inhibition of Cav1.2 [61,62][49][50]. Activation of PKC by phorbol esters and by Gq-coupled receptors also potentiates vascular Cav1.2 activity [63,64,65,66,67,68,69,70][51][52][53][54][55][56][57][58]. Basal Cav1.2 activity is evidently under the tonic control of PKC as PKC inhibition/PKCα depletion enhances Cav1.2 activity in vascular smooth muscle cells [67][55]. Activation of PKG by nitric oxide (NO) suppresses Cav1.2 activity [49][37]. Similar to PKA, PKC is anchored by AKAP150 to adjacent Cav1.2 to alter channel activity [71][59]. Protein tyrosine kinase c-Src promotes tyrosine phosphorylation of the α1c subunit and enhances Cav1.2 activity, which is believed to participate in regulating smooth muscle contractility [72][60]. c-Src via its SH2 and SH3 domains binds to the COOH-terminus of the α1c subunit [70][58]. Y2122 in the COOH-terminus of the α1c subunit appears to be the major phosphorylation site of c-Src [70,73][58][61]. In vascular smooth muscle cells, c-Src enhances Cav1.2 activity [64,74,75,76][52][62][63][64]. Phosphoinositide 3-kinases (PI3Ks) are found to increase Cav1.2 activity in vascular smooth muscle cells [77,78][65][66]. PI3Kγ potentiates Cav1.2 activity by facilitating plasma membrane translocation of α1c subunits and this effect is mediated by AKT/PKB-induced β2 subunit phosphorylation [79,80,81,82][67][68][69][70]. Integrins also participate in the mechanotransduction process of pressure-induced myogenic tone [83][71]. Integrin receptor activation is found to increase Cav1.2 activity via c-Src and PKA [84,85,86][72][73][74].2.2.3. Regulation by Small GTPases
The Ras superfamily of small GTPases (also known as small G-proteins) are cellular switches that regulate a variety of biological processes in living cells. They have been implicated in regulating Ca2+ homeostasis and Cav1.2 is recognized as an important effector of the RGK subfamily (Rem, Rem2, Rad, and Gem/Kir) [87,88][75][76]. Unlike vascular Cav1.2, direct phosphorylation of the cardiac Cav1.2 α1c subunit does not contribute to PKA-stimulated channel activity as mutating all PKA consensus sites in the α1c subunit fails to block the increase in Cav1.2 activity in response to β-adrenergic stimulation [89,90][77][78]. Cardiac Cav1.2 is under tonic inhibition of Rad due to its association to both α1c and/or β subunits [91,92][79][80]. β-adrenergic stimulation induces RAD phosphorylation, promotes the release of RAD from Cav1.2, and increase channel activity [90][78].2.3. Ca
v
1.2 and Myogenic Tone
Vascular smooth muscle cells in resistant arteries and arterioles possess intrinsic ability to contract in response to an increase in intraluminal pressure and to relax upon a decrease in intraluminal pressure [97][81]. This phenomenon is defined as myogenic response and the steady-state of vascular smooth muscle cell contractility in these vessels is termed as myogenic tone. Myogenic tone sets the basal vascular tone and distribution of blood flow to and within tissues/organs. In principle, peripheral vascular resistance can be described by the Poiseuille equation: R = 8Lη/πr4, where R is the resistance, L is length of the vessel, η is viscosity of blood, and r is the radius of the vessel. According to the Poiseuille’s law, peripheral vascular resistance is inversely proportional to the radius to the fourth power. Vascular smooth muscle cells in resistance arteries/arterioles become depolarized in response to increased intraluminal pressure [98,99,100][82][83][84]. Various mechanisms have been proposed to regulate myogenic tone. It is widely believed that pressure-induced membrane depolarization in vascular smooth muscle cells is instigated by mechanosensitive or stretch-activated cation channels including transient receptor potential (TRP) channels and epithelial Na+ channels (ENaCs) [101,102,103,104,105,106,107][85][86][87][88][89][90][91]. The membrane depolarization leads to increased [Ca2+]i and vasoconstriction [99,108][83][92]. The increases in both [Ca2+]i and/or myogenic tone are blocked by the removal of extracellular Ca2+ or by Cav1.2 blockers [99,109,110,111,112,113,114,115][83][93][94][95][96][97][98][99]. These findings suggest that altered intraluminal pressure initiates myogenic tone via membrane depolarization and subsequent opening of Cav1.2. This notion is corroborated by findings from the genetic deletion of Cav1.2 α1c subunit in smooth muscle [116][100]. Such a manipulation abolishes myogenic reactivity in murine tibialis arteries [116][100].3. Roles of Cav1.2 in the Pathogenesis of Hypertension-Related Disorders
3.1. Aberrant Vascular Tone in Hypertension-Related Disorders
Hypertension is associated with increased peripheral vascular resistance. As myogenic tone is the fundamental element of vascular tone, it is reasonable to speculate that myogenic tone is altered in hypertension-related disorders. In patients with hypertension, myogenic tone is increased in coronary arterioles [110][94]. Increased myogenic tone is also noted in resistance arteries from the adipose tissue of paravertebral muscles of hypertensive patients [130][101]. Commonly used rat models of experimental hypertension include the spontaneously hypertensive rat (SHR), stroke-prone spontaneously hypertensive rat (SHRSP), Milan hypertensive strain (MHS), vasopressin-deficient (Di/H) rats, two-kidney, one-clip (2K1C rats), and among others. Compared to the Wistar-Kyoto rat (WKY), myogenic tone of afferent arterioles and arcuate arteries from SHR kidneys is increased [131,132][102][103]. Mesenteric arteries of SHR and MSH also exhibits higher myogenic tone than those of WKY and Milan normotensive strain (MNS), respectively [133,134][104][105]. Elevated myogenic tone is observed in cerebral arteries/basilar arteries of SHR/SHRSP/Di/H compared to WKY and Di normotensive (Di/N) rats, respectively [135,136,137,138][106][107][108][109]. Myogenic tone is increased in skeletal muscle resistance arteries of diabetic patients [144][110]. Various animal models such as type I diabetic rodents induced by streptozotocin (STZ), obese/type II diabetic rodents induced by high-fat diet (HFD), type II diabetic HFD/STZ rodents, type II diabetic Goto-Kakizaki (GK) rats, and type II diabetic C57BL/KsJ-db/db mouse have been developed in diabetes research [145,146][111][112]. Systemic vascular resistance (also known as total vascular resistance) and arterial blood pressure are increased in C57BL/KsJ-db/db mice, HFD rats, and GK rats [147,148,149,150,151,152,153,154,155][113][114][115][116][117][118][119][120][121]. Mesenteric arteries of HFD mice, HFD/STZ mice, and diabetic (db/db) mice displayed higher myogenic tone compared to their counterparts [58,144,155,156][46][110][121][122].3.2. Dysfunction of Vascular Ca
v
1.2 in Hypertension-Related Disorders
As aforementioned, activation of Cav1.2 in vascular smooth muscle cells is essential for the development of myogenic tone [99,100,109,110,113,114,116][83][84][93][94][97][98][100]. The enhanced myogenic tone in peripheral resistance arteries and arterioles in hypertension-related disorders suggests potential dysfunction of vascular Cav1.2. Indeed, this notion is substantiated by lines of evidence from functional studies. First, the increased myogenic tone in resistance arteries/arterioles of hypertensive and diabetic animals was normalized by the Cav1.2 blocker nifedipine [58,132,181][46][103][123]. Whereas specific deletion of the mineralocorticoid receptor reduces KCl- and Cav1.2 agonist Bay K 8644-induced vasoconstriction of mesenteric arteries [142[124][125],182], Bay K 8644-induced contraction and increase in [Ca2+]i in 2K1C rat aorta is greater than in the control 2K rats [183][126]. Animal models of hypertension-related disorders have provided mechanistic insights into the understanding of the Cav1.2 dysfunction in these disorders. Both aberrant expression of and dysregulation of Cav1.2 contribute to vascular Cav1.2 dysfunction. Various studies reveal increased protein expression of α1c [37[28][29][127][128][129],38,187,192,193], α2δ1 [37[28][128],192], and β3 [37,38][28][29] subunits in mesenteric, femoral, and cerebral arteries of SHR and angiotensin II-infused C57BL/6 mice. Consistently, increased expression of Cav1.2 is associated with enhanced channel activity in vascular smooth muscle cells [67,185,193,194,195,196,197][55][129][130][131][132][133][134]. The expression and activity of Cav1.2 are reduced in mesenteric arteries of aged mice lacking mineralocorticoid receptors in smooth muscle cells [143][135]. Dexamethasone administration increases the expression of the cardiac Cav1.2 α1c subunit in rats [198][136]. As expected, Cav1.2 activity in A7r5 cells is increased following chronic dexamethasone exposure [199][137]. An increase in protein expression of the α1c subunits is also observed in cerebral arteries of STZ rats, which is associated with increased Cav1.2 activity and contraction to KCl [203][138]. Cav1.2 also displays increased activity in vascular smooth muscle cells of cerebral and mesenteric arteries from STZ, GK, and HFD rats and db/db mice [153,204,205,206][119][139][140][141]. Human arteries from diabetic patients have higher Cav1.2 activity due to increased phosphorylation of α1C at Ser1928 by PKA [56][44]. Gal-1 is highly expressed in the placenta [207][142]. Circulating Gal-1 level also increases during gestation [208][143]. Given the critical role of Gal-1 in regulating Cav1.2 surface expression/activity discussed above, it is reasonable to speculate that the elevated Gal-1 in pregnancy may contribute to reduced uterine arterial myogenic tone by suppressing Cav1.2 surface expression/activity [209,210][144][145].4. Roles of Reactive Oxygen Species (ROS) in the Pathogenesis of Hypertensive Disorders
4.1. Overview of ROS
Reactive oxygen species (ROS) are oxygen-containing molecules naturally produced in cellular metabolism. ROS comprise free radicals such as superoxide anion (O2•−) and hydroxyl radical (•OH) and nonradical molecule hydrogen peroxide (H2O2). O2•− is formed from the one-electron reduction of molecular oxygen (O2). It is a precursor to a cascade of other ROS. Its dismutation, either occurring spontaneously or being catalyzed by superoxide dismutases (SODs), produces H2O2. Through the Fenton reaction, H2O2 can be reduced to •OH. Moreover, the reaction between O2•− and nitric oxide (NO) results in the formation of peroxynitrite (ONOO−). ROS are produced in different cellular compartments including mitochondria, endoplasmic reticulum (ER), lysosomes, peroxisomes, and plasma membrane [23,213,214][14][146][147]. Nicotinamide adenine dinucleotide phosphate oxidases (NOXs) and mitochondria are the major sources of ROS [215][148] (Figure 32). NOXs are a family of transmembrane proteins that catalyze the transfer of electron donated by NADPH to molecular oxygen to form O2•− [216][149]. NOXs comprise seven isoforms (NOX1-5 and dual oxidases (DUOX) 1 and 2). NOX1, NOX2, NOX4, and NOX5 are primary isoforms in vasculature [217][150].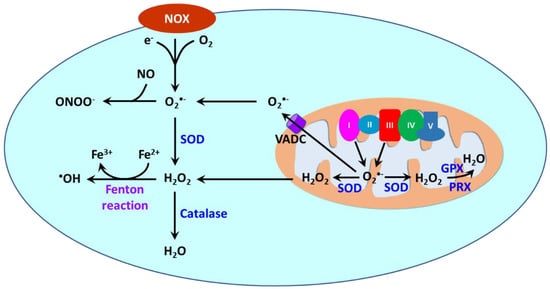
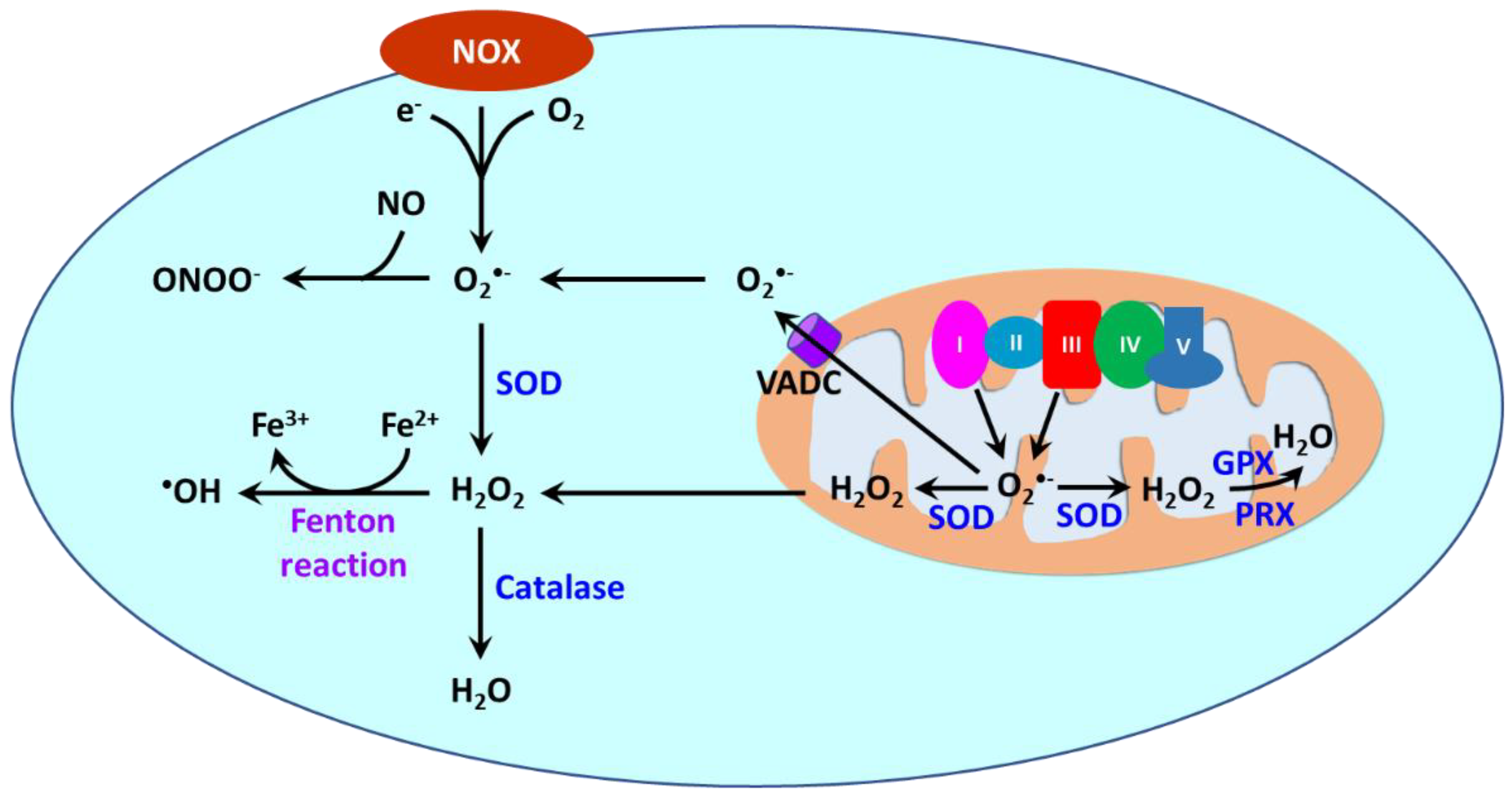
4.2. Oxidative Stress as a Hallmark in Hypertension-Related Disorders
4.2.1. NOX-Derived ROS in Hypertension-Related Disorders
Several polymorphisms in the gene encoding p22phox in human are associated with hypertension by affecting enzymatic activity [310][176]. In SHR/SHRSP, the expression/activity of vascular NOXs 1, 2 and 4 are increased [270,272,311,312,313,314,315,316][177][178][179][180][181][182][183][184]. DOCA rats have higher vascular expression of NOX subunit p22phox and enzymatic activity [270,317][177][185]. NOX activity in cultured vascular smooth cells and in cultured rat mesangial cells is also stimulated by aldosterone, leading to increased ROS generation [318,319][186][187]. Chronic administration of aldosterone causes NOX2-depednet increase O2•− production in mouse cerebral arteries [277][188]. Dexamethasone upregulates Nox1 expression in rat aorta via activation of the glucocorticoid receptor [320][189]. In a mouse model of hyperadrenergic hypertension created by targeted ablation of the chromogranin a (Chga) gene, renal expression of NOX1/2 is increased [321][190]. Angiotensin II-infused rodents are associated with elevated expression of NOXs 1 and 2 as well as NOX subunits p22phox, p47phox, and Rac1 in vessels and increased NOX activity [268,274,322,323,324,325,326,327][191][192][193][194][195][196][197][198]. Angiotensin II apparently contributes to the upregulation of NOXs as it stimulates gp91phox (NOX2) and p22phox expression in vascular smooth muscle cells from human resistance arteries [328][199]. In diabetic patients, vascular protein abundance of NOX subunits p22phox, p67phox, and p47phox and enzymatic activity of NOXs are increased [279][200]. Moreover, O2•− production is reduced by non-selective NOX inhibitor diphenylene iodonium in vessels from diabetic patients [279][200]. The expression and activity of NOXs 1, 2 and 4 as well as p22phox/p47phox are increased in aorta and mesenteric arteries of STZ rodents [287,308,329,330,331,332,333][201][202][203][204][205][206][207]. Vascular expression/activity of NOXs 1, 2, and 4 as well as p22phox are also elevated in db/db mice [280,334,335][208][209][210]. Chronic high glucose exposure stimulates p22phox, p47phox and p67phox expression and NOX activity in cultured endothelial cells [285,286,336,337,338][211][212][213][214][215].4.2.2. Mitochondria-Derived ROS in Hypertension-Related Disorders
Mitochondria become dysfunctional in hypertension-related disorders. SIRT3, a histone deacetylase, is an important regulator of mitochondrial redox state. It interacts with SOD2 in mitochondria and subsequently promotes SOD2 deacetylation, leading to enhanced enzymatic activity [355,356][216][217]. Essential hypertension in human is associated with increased mitochondrial oxidative stress in arterioles from mediastinal fat due to reduced SIRT3 and increased SOD2 acetylation [357][218]. Similar to human essential hypertension, SHR also exhibits overproduction of mitochondrial ROS in vessels [358][219]. Partial deletion of SOD2 in mitochondria (SOD2+/−) increases renal oxidative stress and blood pressure [359][220]. In angiotensin II-infused mice, the increased blood pressure is associated with increased SIRT3 S-glutathionylation and vascular SOD2 acetylation, reduced SOD2 activity and elevated vascular O2•− production [357,360][218][221]. Those alterations are diminished by SIRT3 overexpression [357][218]. Cyclophilin D is a regulatory subunit of the mitochondrial permeability transition pore and participates in regulating mitochondrial function [361][222]. Evidently, it is an important mediator of angiotensin II-induced hypertension as its depletion diminishes both mitochondrial O2•− generation in aorta and hypertension [362][223]. Mitochondrial O2•− generation in DOCA rat mesenteric arteries is also increased [255][224]. In cultured HUVECs, excess aldosterone suppresses SOD2 expression and increases mitochondrial ROS production [363][225]. Elevation of mitochondrial O2•− is detected in subcutaneous arterioles in type 2 diabetic patients [366][226]. Likewise, mitochondrial H2O2 is increased in primary human saphenous vein endothelial cells from type 2 diabetic subjects [367][227]. STZ mice/rats, HFD mice, ZDF rats, and GK rats display increased mitochondrial ROS in vascular smooth muscle cells and endothelial cells [331,368,369,370,371,372][205][228][229][230][231][232]. Hyperglycemia is apparently a causative factor leading to heightened mitochondrial ROS in vascular cells. Exposure to high concentrations of glucose stimulates mitochondrial ROS production in a variety of endothelial cells, promoting mitochondrial damage by producing 8-hydroxydeoxyguanosine and nitrotyrosine [373,374,375,376,377,378,379,380,381][233][234][235][236][237][238][239][240][241].4.3. A Causative Role of ROS in in Animal Models of Hypertension-Related Disorders
Oxidative stress is believed to be linked to pathogenesis and progression of a myriad of human diseases including hypertension-related disorders [21,23,24,397,398,399,400,401][12][14][15][242][243][244][245][246]. Numerous studies using animal models reveal a causative role of ROS in the pathogenesis of hypertension, diabetes, and preeclampsia. Treatment with the SOD mimetic tempol reduces vascular ROS and lower blood pressure in animal models of essential and secondary hypertension [253,257,269,317,402[185][247][248][249][250][251][252],403,404], of diabetes [405[253][254][255][256],406,407,408], and of preeclampsia [293,294,346,409][257][258][259][260]. SOD1 deletion promotes development of hypertension, whereas the delivery of liposome-encapsulated SOD diminishes Ang II-induced hypertension [267,410][261][262]. NOX inhibitors apocynin and diphenyleneiodonium have been shown to decrease vascular ROS, improve vascular function, and lower blood pressure in animal models of hypertension-related disorders [272,275,312,317,324,340,411,412,413,414,415][178][180][185][195][263][264][265][266][267][268][269]. Angiotensin II-induced vascular ROS and hypertension is enhanced in transgenic mice overexpressing NOX1 in smooth muscle cells and is suppressed in NOX1 deficient mice [274,325,416][192][196][270].5. Contribution of Dysfunctional Cav1.2 Conferred by ROS to Increased Vascular Tone in Hypertension-Related Disorders
5.1. ROS and Ca
v
1.2 Function/Expression
There is ample evidence that Cav1.2 is regulated by cellular redox state. The α1c subunit contains 48 cysteines with some of them being sensitive to redox modulation [420][271]. Redox modulation of sulfhydryl groups of cysteine residues could alter the structure and function of proteins. Site-directed mutagenesis has identified several cysteine residues including C543, C1789, C1790, and C1810 that subject to redox regulation [421,422][272][273]. However, electrophysiological studies reveal conflicting effects of ROS on Cav1.2. Studies from Amberg’s group demonstrate that bath application of H2O2 and ROS produced by xanthine oxidase/hypoxanthine stimulates Cav1.2 activity in rat cerebral arterial smooth muscle cells [423,424][274][275]. Both mitochondria- and NOX-derived ROS could exert stimulatory effects on Cav1.2. Antimycin enhances Cav1.2 activity by promoting mitochondrial H2O2 generation. The redox modulation of Cav1.2 has immense impacts on vascular function. H2O2 is reported to trigger an increase in [Ca2+]i and vasoconstriction in rat mesenteric arteries, rat coronary arteries, rat aorta, and canine cerebral arteries [429,430,431,432,433,434,435][276][277][278][279][280][281][282]. H2O2-induced increase in [Ca2+]i and contractile response are inhibited by the removal of extracellular Ca2+ and by Cav1.2 inhibitors nisoldipine, nifedipine, diltiazem, and verapamil, suggesting that H2O2 activates Cav1.2 in vascular smooth muscle cells [429,430,431,434][276][277][278][281]. NO is also a reactive, gaseous signaling molecule. S-nitrosylation, the covalent attachment of NO to a cysteine thiol in a given protein, is also an important redox signaling [445][283]. S-nitrosylation of Cys 1180 and/or Cys1280 in Cav1.2 reduces surface expression of the channel and channel activity [446][284]. In addition, S-nitrosylation of Cav1.2 also reduces channel open probability [447][285]. Excess ROS leads to low NO bioavailability in hypertension [448][286]. Remarkably, ROS also regulate gene expression [460,461][287][288]. There is a concurring decrease in both vascular ROS and Cav1.2 expression in aortas of aged mice lacking the mineralocorticoid receptor in smooth muscle cells [142][124]. The protein expression of the α1C subunit is increased by angiotensin II in cultured rat mesenteric arteries [462,463][289][290]. H2O2 mimics, while NOX inhibition by apocynin, diphenyleneiodonium and gp91ds-tat as well as catalase annuls angiotensin II-mediated upregulation of the α1C subunit [463][290]. In a cardiac muscle cell line HL-1 cells, angiotensin II upregulates the α1C subunit at both mRNA and protein levels through the NOX-PKC pathway-mediated cAMP response element binding protein (CREB) phosphorylation [464][291]. It is expected that this mechanism could also play a role in secondary hypertension as hypertension due to Cushing’s syndrome, hyperaldosteronism, and renovascular disease involves activation of the renin-angiotensin system [231,256,465,466,467,468,469][158][292][293][294][295][296][297].5.2. ROS and Myogenic Tone
Exogenous ROSs have been shown to alter myogenic tone. In pressurized mouse tail arterioles and rat gracilis skeletal muscle arterioles, H2O2 causes vasoconstriction [471,472][298][299]. The exposure of rat cerebral arteries to the ROS-generating system xanthine oxidase plus hypoxanthine also increases myogenic tone [423][274]. Likewise, O2•− generated by paraquat enhances myogenic tone in mouse afferent arterioles [473][300]. •OH generated by the Fenton reaction from H2O2 and the iron redox chelate Fe3+/nitrilotriacetate (FeNTA) elevates myogenic tone in denuded rat ophthalmic arteries [474][301]. Numerous studies demonstrate that PKC/c-Src activation contributes to myogenic tone development in small arteries/arterioles [136,487,488,489,490,491][107][302][303][304][305][306]. The expression/activity of vascular PKC/c-Src is increased in SHR and STZ rats as well as in uterine arteries of high-altitude pregnant sheep [179,435,492,493][282][307][308][309]. PKC/c-Src activation has a greater contribution to myogenic tone in SHR cerebral arteries, STZ rat gracilis skeletal muscle arterioles, and high-altitude sheep uterine arteries [136,159,179][107][307][310].6. Conclusions
Evidently, Ca2+ influx through Cav1.2 is fundamental to vasoconstriction and myogenic tone of small arteries and arterioles. Antihypertensive drugs including Cav1.2 blockers have been successfully used to the management of hypertension-related disorders [495,496,497,498][311][312][313][314]. Oxidative stress is a hallmark of hypertension-related disorders. However, it is still an open question whether oxidative stress is a cause or consequence of hypertension. The data highlights critical roles of vascular oxidative stress in the pathophysiology of hypertension-related disorders. Vascular Cav1.2 is targeted by excessive ROS directly and indirectly leading to exaggerated channel expression/activity. The dysfunction of Cav1.2 ultimately results in increased myogenic tone and elevated blood pressure. In preclinical studies, ROS-induced myogenic tone is diminished by Cav1.2 blocker nifedipine [473][300].References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Woll, K.A.; Van Petegem, F. Calcium-release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022, 102, 209–268.
- Ghosh, D.; Syed, A.U.; Prada, M.P.; Nystoriak, M.A.; Santana, L.F.; Nieves-Cintron, M.; Navedo, M.F. Calcium Channels in Vascular Smooth Muscle. Adv. Pharmacol. 2017, 78, 49–87.
- Ottolini, M.; Hong, K.; Sonkusare, S.K. Calcium signals that determine vascular resistance. Wiley Interdiscip Rev. Syst. Biol. Med. 2019, 11, e1448.
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324.
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis, and Management Recommendations for International Practice. Hypertension 2018, 72, 24–43.
- Justin, J.; Fayol, A.; Bruno, R.M.; Khettab, H.; Boutouyrie, P. International Guidelines for Hypertension: Resemblance, Divergence and Inconsistencies. J. Clin. Med. 2022, 11, 1975.
- de Boer, I.H.; Bangalore, S.; Benetos, A.; Davis, A.M.; Michos, E.D.; Muntner, P.; Rossing, P.; Zoungas, S.; Bakris, G. Diabetes and Hypertension: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 1273–1284.
- Tsimihodimos, V.; Gonzalez-Villalpando, C.; Meigs, J.B.; Ferrannini, E. Hypertension and Diabetes Mellitus: Coprediction and Time Trajectories. Hypertension 2018, 71, 422–428.
- Shin, D.; Bohra, C.; Kongpakpaisarn, K. Impact of the Discordance Between the American College of Cardiology/American Heart Association and American Diabetes Association Recommendations on Hypertension in Patients With Diabetes Mellitus in the United States. Hypertension 2018, 72, 256–259.
- Michael, S.K.; Surks, H.K.; Wang, Y.; Zhu, Y.; Blanton, R.; Jamnongjit, M.; Aronovitz, M.; Baur, W.; Ohtani, K.; Wilkerson, M.K.; et al. High blood pressure arising from a defect in vascular function. Proc. Natl. Acad. Sci. USA 2008, 105, 6702–6707.
- Raijmakers, M.T.; Dechend, R.; Poston, L. Oxidative stress and preeclampsia: Rationale for antioxidant clinical trials. Hypertension 2004, 44, 374–380.
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668.
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 2015, 31, 631–641.
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539.
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 2016, 594, 5369–5390.
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947.
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102.
- Kobrinsky, E.; Tiwari, S.; Maltsev, V.A.; Harry, J.B.; Lakatta, E.; Abernethy, D.R.; Soldatov, N.M. Differential role of the alpha1C subunit tails in regulation of the Cav1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J. Biol. Chem. 2005, 280, 12474–12485.
- Campiglio, M.; Flucher, B.E. The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell. Physiol. 2015, 230, 2019–2031.
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425.
- Mikami, A.; Imoto, K.; Tanabe, T.; Niidome, T.; Mori, Y.; Takeshima, H.; Narumiya, S.; Numa, S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 1989, 340, 230–233.
- Biel, M.; Ruth, P.; Bosse, E.; Hullin, R.; Stuhmer, W.; Flockerzi, V.; Hofmann, F. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 1990, 269, 409–412.
- Murakami, M.; Yamamura, H.; Suzuki, T.; Kang, M.G.; Ohya, S.; Murakami, A.; Miyoshi, I.; Sasano, H.; Muraki, K.; Hano, T.; et al. Modified cardiovascular L-type channels in mice lacking the voltage-dependent Ca2+ channel beta3 subunit. J. Biol. Chem. 2003, 278, 43261–43267.
- Bannister, J.P.; Adebiyi, A.; Zhao, G.; Narayanan, D.; Thomas, C.M.; Feng, J.Y.; Jaggar, J.H. Smooth muscle cell alpha2delta-1 subunits are essential for vasoregulation by Cav1.2 channels. Circ. Res. 2009, 105, 948–955.
- Cox, R.H.; Fromme, S. Expression of Calcium Channel Subunit Variants in Small Mesenteric Arteries of WKY and SHR. Am. J. Hypertens. 2015, 28, 1229–1239.
- Kharade, S.V.; Sonkusare, S.K.; Srivastava, A.K.; Thakali, K.M.; Fletcher, T.W.; Rhee, S.W.; Rusch, N.J. The beta3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension 2013, 61, 137–142.
- Chen, Y.H.; Li, M.H.; Zhang, Y.; He, L.L.; Yamada, Y.; Fitzmaurice, A.; Shen, Y.; Zhang, H.; Tong, L.; Yang, J. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 2004, 429, 675–680.
- Jay, S.D.; Sharp, A.H.; Kahl, S.D.; Vedvick, T.S.; Harpold, M.M.; Campbell, K.P. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J. Biol. Chem. 1991, 266, 3287–3293.
- Cheng, X.; Pachuau, J.; Blaskova, E.; Asuncion-Chin, M.; Liu, J.; Dopico, A.M.; Jaggar, J.H. Alternative splicing of Cav1.2 channel exons in smooth muscle cells of resistance-size arteries generates currents with unique electrophysiological properties. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H680–H688.
- Cheng, X.; Liu, J.; Asuncion-Chin, M.; Blaskova, E.; Bannister, J.P.; Dopico, A.M.; Jaggar, J.H. A novel Cav1.2 N terminus expressed in smooth muscle cells of resistance size arteries modifies channel regulation by auxiliary subunits. J. Biol. Chem. 2007, 282, 29211–29221.
- Puri, T.S.; Gerhardstein, B.L.; Zhao, X.L.; Ladner, M.B.; Hosey, M.M. Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry 1997, 36, 9605–9615.
- Gerhardstein, B.L.; Puri, T.S.; Chien, A.J.; Hosey, M.M. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the beta 2 subunit of L-type voltage-dependent calcium channels. Biochemistry 1999, 38, 10361–10370.
- Bunemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J. Biol. Chem. 1999, 274, 33851–33854.
- Yang, L.; Liu, G.; Zakharov, S.I.; Bellinger, A.M.; Mongillo, M.; Marx, S.O. Protein kinase G phosphorylates Cav1.2 alpha1c and beta2 subunits. Circ. Res. 2007, 101, 465–474.
- Li, Y.; Yang, H.; He, T.; Zhang, L.; Liu, C. Post-Translational Modification of Cav1.2 and its Role in Neurodegenerative Diseases. Front. Pharmacol. 2021, 12, 775087.
- Ishikawa, T.; Hume, J.R.; Keef, K.D. Regulation of Ca2+ channels by cAMP and cGMP in vascular smooth muscle cells. Circ. Res. 1993, 73, 1128–1137.
- Liu, H.; Xiong, Z.; Sperelakis, N. Cyclic nucleotides regulate the activity of L-type calcium channels in smooth muscle cells from rat portal vein. J. Mol. Cell. Cardiol. 1997, 29, 1411–1421.
- Ruiz-Velasco, V.; Zhong, J.; Hume, J.R.; Keef, K.D. Modulation of Ca2+ channels by cyclic nucleotide cross activation of opposing protein kinases in rabbit portal vein. Circ. Res. 1998, 82, 557–565.
- Keef, K.D.; Hume, J.R.; Zhong, J. Regulation of cardiac and smooth muscle Ca2+ channels (Cav1.2a,b) by protein kinases. Am. J. Physiol. Cell Physiol. 2001, 281, C1743–C1756.
- Fusi, F.; Manetti, F.; Durante, M.; Sgaragli, G.; Saponara, S. The vasodilator papaverine stimulates L-type Ca2+ current in rat tail artery myocytes via a PKA-dependent mechanism. Vasc. Pharmacol. 2016, 76, 53–61.
- Nystoriak, M.A.; Nieves-Cintron, M.; Patriarchi, T.; Buonarati, O.R.; Prada, M.P.; Morotti, S.; Grandi, E.; Fernandes, J.D.; Forbush, K.; Hofmann, F.; et al. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel Cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 2017, 10, eaaf9647.
- Fusi, F.; Mugnai, P.; Trezza, A.; Spiga, O.; Sgaragli, G. Fine tuning by protein kinases of Cav1.2 channel current in rat tail artery myocytes. Biochem. Pharmacol. 2020, 182, 114263.
- Syed, A.U.; Reddy, G.R.; Ghosh, D.; Prada, M.P.; Nystoriak, M.A.; Morotti, S.; Grandi, E.; Sirish, P.; Chiamvimonvat, N.; Hell, J.W.; et al. Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J. Clin. Investig. 2019, 129, 3140–3152.
- Xiong, Z.; Sperelakis, N.; Fenoglio-Preiser, C. Regulation of L-type calcium channels by cyclic nucleotides and phosphorylation in smooth muscle cells from rabbit portal vein. J. Vasc. Res. 1994, 31, 271–279.
- Taguchi, K.; Ueda, M.; Kubo, T. Effects of cAMP and cGMP on L-type calcium channel currents in rat mesenteric artery cells. Jpn. J. Pharmacol. 1997, 74, 179–186.
- Quignard, J.F.; Frapier, J.M.; Harricane, M.C.; Albat, B.; Nargeot, J.; Richard, S. Voltage-gated calcium channel currents in human coronary myocytes. Regulation by cyclic GMP and nitric oxide. J. Clin. Investig. 1997, 99, 185–193.
- Sharma, N.; Bhattarai, J.P.; Hwang, P.H.; Han, S.K. Nitric oxide suppresses L-type calcium currents in basilar artery smooth muscle cells in rabbits. Neurol. Res. 2013, 35, 424–428.
- Schuhmann, K.; Groschner, K. Protein kinase-C mediates dual modulation of L-type Ca2+ channels in human vascular smooth muscle. FEBS Lett. 1994, 341, 208–212.
- Callaghan, B.; Koh, S.D.; Keef, K.D. Muscarinic M2 receptor stimulation of Cav1.2b requires phosphatidylinositol 3-kinase, protein kinase C, and c-Src. Circ. Res. 2004, 94, 626–633.
- Navedo, M.F.; Amberg, G.C.; Votaw, V.S.; Santana, L.F. Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA 2005, 102, 11112–11117.
- Cobine, C.A.; Callaghan, B.P.; Keef, K.D. Role of L-type calcium channels and PKC in active tone development in rabbit coronary artery. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3079–H3088.
- Nieves-Cintron, M.; Amberg, G.C.; Navedo, M.F.; Molkentin, J.D.; Santana, L.F. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc. Natl. Acad. Sci. USA 2008, 105, 15623–15628.
- Ren, C.; Zhang, J.; Philipson, K.D.; Kotlikoff, M.I.; Blaustein, M.P.; Matteson, D.R. Activation of L-type Ca2+ channels by protein kinase C is reduced in smooth muscle-specific Na+/Ca2+ exchanger knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1484–H1491.
- Weiss, S.; Keren-Raifman, T.; Oz, S.; Ben Mocha, A.; Haase, H.; Dascal, N. Modulation of distinct isoforms of L-type calcium channels by G(q)-coupled receptors in Xenopus oocytes: Antagonistic effects of Gbetagamma and protein kinase C. Channels 2012, 6, 426–437.
- Gulia, J.; Navedo, M.F.; Gui, P.; Chao, J.T.; Mercado, J.L.; Santana, L.F.; Davis, M.J. Regulation of L-type calcium channel sparklet activity by c-Src and PKC-alpha. Am. J. Physiol. Cell Physiol. 2013, 305, C568–C577.
- Navedo, M.F.; Nieves-Cintron, M.; Amberg, G.C.; Yuan, C.; Votaw, V.S.; Lederer, W.J.; McKnight, G.S.; Santana, L.F. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ. Res. 2008, 102, e1–e11.
- Hu, X.Q.; Singh, N.; Mukhopadhyay, D.; Akbarali, H.I. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J. Biol. Chem. 1998, 273, 5337–5342.
- Gui, P.; Chao, J.T.; Wu, X.; Yang, Y.; Davis, G.E.; Davis, M.J. Coordinated regulation of vascular Ca2+ and K+ channels by integrin signaling. Adv. Exp. Med. Biol. 2010, 674, 69–79.
- Wijetunge, S.; Hughes, A.D. pp60c-src increases voltage-operated calcium channel currents in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1995, 217, 1039–1044.
- Wijetunge, S.; Hughes, A.D. Activation of endogenous c-Src or a related tyrosine kinase by intracellular (pY)EEI peptide increases voltage-operated calcium channel currents in rabbit ear artery cells. FEBS Lett. 1996, 399, 63–66.
- Wijetunge, S.; Lymn, J.S.; Hughes, A.D. Effects of protein tyrosine kinase inhibitors on voltage-operated calcium channel currents in vascular smooth muscle cells and pp60(c-src) kinase activity. Br. J. Pharmacol. 2000, 129, 1347–1354.
- Macrez, N.; Mironneau, C.; Carricaburu, V.; Quignard, J.F.; Babich, A.; Czupalla, C.; Nurnberg, B.; Mironneau, J. Phosphoinositide 3-kinase isoforms selectively couple receptors to vascular L-type Ca2+ channels. Circ. Res. 2001, 89, 692–699.
- Pinho, J.F.; Medeiros, M.A.; Capettini, L.S.; Rezende, B.A.; Campos, P.P.; Andrade, S.P.; Cortes, S.F.; Cruz, J.S.; Lemos, V.S. Phosphatidylinositol 3-kinase-delta up-regulates L-type Ca2+ currents and increases vascular contractility in a mouse model of type 1 diabetes. Br. J. Pharmacol. 2010, 161, 1458–1471.
- Le Blanc, C.; Mironneau, C.; Barbot, C.; Henaff, M.; Bondeva, T.; Wetzker, R.; Macrez, N. Regulation of vascular L-type Ca2+ channels by phosphatidylinositol 3,4,5-trisphosphate. Circ. Res. 2004, 95, 300–307.
- Viard, P.; Butcher, A.J.; Halet, G.; Davies, A.; Nurnberg, B.; Heblich, F.; Dolphin, A.C. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 2004, 7, 939–946.
- Catalucci, D.; Zhang, D.H.; DeSantiago, J.; Aimond, F.; Barbara, G.; Chemin, J.; Bonci, D.; Picht, E.; Rusconi, F.; Dalton, N.D.; et al. Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability. J. Cell Biol. 2009, 184, 923–933.
- Carnevale, D.; Vecchione, C.; Mascio, G.; Esposito, G.; Cifelli, G.; Martinello, K.; Landolfi, A.; Selvetella, G.; Grieco, P.; Damato, A.; et al. PI3Kgamma inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc. Res. 2012, 93, 200–209.
- Davis, M.J.; Wu, X.; Nurkiewicz, T.R.; Kawasaki, J.; Davis, G.E.; Hill, M.A.; Meininger, G.A. Integrins and mechanotransduction of the vascular myogenic response. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1427–H1433.
- Wu, X.; Davis, G.E.; Meininger, G.A.; Wilson, E.; Davis, M.J. Regulation of the L-type calcium channel by alpha 5beta 1 integrin requires signaling between focal adhesion proteins. J. Biol. Chem. 2001, 276, 30285–30292.
- Gui, P.; Wu, X.; Ling, S.; Stotz, S.C.; Winkfein, R.J.; Wilson, E.; Davis, G.E.; Braun, A.P.; Zamponi, G.W.; Davis, M.J. Integrin receptor activation triggers converging regulation of Cav1.2 calcium channels by c-Src and protein kinase A pathways. J. Biol. Chem. 2006, 281, 14015–14025.
- Waitkus-Edwards, K.R.; Martinez-Lemus, L.A.; Wu, X.; Trzeciakowski, J.P.; Davis, M.J.; Davis, G.E.; Meininger, G.A. alpha(4)beta(1) Integrin activation of L-type calcium channels in vascular smooth muscle causes arteriole vasoconstriction. Circ. Res. 2002, 90, 473–480.
- Correll, R.N.; Pang, C.; Niedowicz, D.M.; Finlin, B.S.; Andres, D.A. The RGK family of GTP-binding proteins: Regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell. Signal. 2008, 20, 292–300.
- Yang, T.; Colecraft, H.M. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta 2013, 1828, 1644–1654.
- Katchman, A.; Yang, L.; Zakharov, S.I.; Kushner, J.; Abrams, J.; Chen, B.X.; Liu, G.; Pitt, G.S.; Colecraft, H.M.; Marx, S.O. Proteolytic cleavage and PKA phosphorylation of alpha1C subunit are not required for adrenergic regulation of Cav1.2 in the heart. Proc. Natl. Acad. Sci. USA 2017, 114, 9194–9199.
- Liu, G.; Papa, A.; Katchman, A.N.; Zakharov, S.I.; Roybal, D.; Hennessey, J.A.; Kushner, J.; Yang, L.; Chen, B.X.; Kushnir, A.; et al. Mechanism of adrenergic Cav1.2 stimulation revealed by proximity proteomics. Nature 2020, 577, 695–700.
- Yang, T.; Puckerin, A.; Colecraft, H.M. Distinct RGK GTPases differentially use alpha1- and auxiliary beta-binding-dependent mechanisms to inhibit Cav1.2/Cav2.2 channels. PLoS ONE 2012, 7, e37079.
- Puckerin, A.A.; Chang, D.D.; Shuja, Z.; Choudhury, P.; Scholz, J.; Colecraft, H.M. Engineering selectivity into RGK GTPase inhibition of voltage-dependent calcium channels. Proc. Natl. Acad. Sci. USA 2018, 115, 12051–12056.
- Davis, M.J.; Hill, M.A. Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 1999, 79, 387–423.
- Harder, D.R. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ. Res. 1984, 55, 197–202.
- Knot, H.J.; Nelson, M.T. Regulation of arterial diameter and wall in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 1998, 508 Pt 1, 199–209.
- Kotecha, N.; Hill, M.A. Myogenic contraction in rat skeletal muscle arterioles: Smooth muscle membrane potential and Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1326–H1334.
- Welsh, D.G.; Morielli, A.D.; Nelson, M.T.; Brayden, J.E. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ. Res. 2002, 90, 248–250.
- Earley, S.; Waldron, B.J.; Brayden, J.E. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ. Res. 2004, 95, 922–929.
- Sharif-Naeini, R.; Dedman, A.; Folgering, J.H.; Duprat, F.; Patel, A.; Nilius, B.; Honore, E. TRP channels and mechanosensory transduction: Insights into the arterial myogenic response. Pflügers Arch. 2008, 456, 529–540.
- Kim, E.C.; Choi, S.K.; Lim, M.; Yeon, S.I.; Lee, Y.H. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS ONE 2013, 8, e84194.
- Nemeth, Z.; Hildebrandt, E.; Ryan, M.J.; Granger, J.P.; Drummond, H.A. Pressure-induced constriction of the middle cerebral artery is abolished in TrpC6 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H42–H50.
- Jackson, W.F. Myogenic Tone in Peripheral Resistance Arteries and Arterioles: The Pressure Is On! Front. Physiol. 2021, 12, 699517.
- Skaik, K.; Shahzad, M. The emerging role of TRPV1 in myogenic tone. J. Physiol. 2022, 600, 2287–2288.
- Nilsson, H.; Jensen, P.E.; Mulvany, M.J. Minor role for direct adrenoceptor-mediated calcium entry in rat mesenteric small arteries. J. Vasc. Res. 1994, 31, 314–321.
- Wesselman, J.P.; VanBavel, E.; Pfaffendorf, M.; Spaan, J.A. Voltage-operated calcium channels are essential for the myogenic responsiveness of cannulated rat mesenteric small arteries. J. Vasc. Res. 1996, 33, 32–41.
- Miller, F.J., Jr.; Dellsperger, K.C.; Gutterman, D.D. Myogenic constriction of human coronary arterioles. Am. J. Physiol. 1997, 273, H257–H264.
- Potocnik, S.J.; Murphy, T.V.; Kotecha, N.; Hill, M.A. Effects of mibefradil and nifedipine on arteriolar myogenic responsiveness and intracellular Ca2+. Br. J. Pharmacol. 2000, 131, 1065–1072.
- Murphy, T.V.; Spurrell, B.E.; Hill, M.A. Mechanisms underlying pervanadate-induced contraction of rat cremaster muscle arterioles. Eur. J. Pharmacol. 2002, 442, 107–114.
- Ahmed, A.; Waters, C.M.; Leffler, C.W.; Jaggar, J.H. Ionic mechanisms mediating the myogenic response in newborn porcine cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2061–H2069.
- Abd El-Rahman, R.R.; Harraz, O.F.; Brett, S.E.; Anfinogenova, Y.; Mufti, R.E.; Goldman, D.; Welsh, D.G. Identification of L- and T-type Ca2+ channels in rat cerebral arteries: Role in myogenic tone development. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H58–H71.
- Jackson, W.F.; Boerman, E.M. Voltage-gated Ca2+ channel activity modulates smooth muscle cell calcium waves in hamster cremaster arterioles. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H871–H878.
- Moosmang, S.; Schulla, V.; Welling, A.; Feil, R.; Feil, S.; Wegener, J.W.; Hofmann, F.; Klugbauer, N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003, 22, 6027–6034.
- Carnevale, D.; Facchinello, N.; Iodice, D.; Bizzotto, D.; Perrotta, M.; De Stefani, D.; Pallante, F.; Carnevale, L.; Ricciardi, F.; Cifelli, G.; et al. Loss of EMILIN-1 Enhances Arteriolar Myogenic Tone Through TGF-beta (Transforming Growth Factor-beta)-Dependent Transactivation of EGFR (Epidermal Growth Factor Receptor) and Is Relevant for Hypertension in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2484–2497.
- Ren, Y.; D’Ambrosio, M.A.; Liu, R.; Pagano, P.J.; Garvin, J.L.; Carretero, O.A. Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1769–H1775.
- Nademi, S.; Lu, C.; Dickhout, J.G. Enhanced Myogenic Constriction in the SHR Preglomerular Vessels Is Mediated by Thromboxane A2 Synthesis. Front. Physiol. 2020, 11, 853.
- Izzard, A.S.; Bund, S.J.; Heagerty, A.M. Myogenic tone in mesenteric arteries from spontaneously hypertensive rats. Am. J. Physiol. 1996, 270, H1–H6.
- Linde, C.I.; Karashima, E.; Raina, H.; Zulian, A.; Wier, W.G.; Hamlyn, J.M.; Ferrari, P.; Blaustein, M.P.; Golovina, V.A. Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H611–H620.
- Dunn, W.R.; Wallis, S.J.; Gardiner, S.M. Remodelling and enhanced myogenic tone in cerebral resistance arteries isolated from genetically hypertensive Brattleboro rats. J. Vasc. Res. 1998, 35, 18–26.
- Jarajapu, Y.P.; Knot, H.J. Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1917–H1922.
- Ahn, D.S.; Choi, S.K.; Kim, Y.H.; Cho, Y.E.; Shin, H.M.; Morgan, K.G.; Lee, Y.H. Enhanced stretch-induced myogenic tone in the basilar artery of spontaneously hypertensive rats. J. Vasc. Res. 2007, 44, 182–191.
- Gonzalez, J.M.; Somoza, B.; Conde, M.V.; Fernandez-Alfonso, M.S.; Gonzalez, M.C.; Arribas, S.M. Hypertension increases middle cerebral artery resting tone in spontaneously hypertensive rats: Role of tonic vasoactive factor availability. Clin. Sci. 2008, 114, 651–659.
- Sauve, M.; Hui, S.K.; Dinh, D.D.; Foltz, W.D.; Momen, A.; Nedospasov, S.A.; Offermanns, S.; Husain, M.; Kroetsch, J.T.; Lidington, D.; et al. Tumor Necrosis Factor/Sphingosine-1-Phosphate Signaling Augments Resistance Artery Myogenic Tone in Diabetes. Diabetes 2016, 65, 1916–1928.
- King, A.; Bowe, J. Animal models for diabetes: Understanding the pathogenesis and finding new treatments. Biochem. Pharmacol. 2016, 99, 1–10.
- Kottaisamy, C.P.D.; Raj, D.S.; Prasanth Kumar, V.; Sankaran, U. Experimental animal models for diabetes and its related complications—A review. Lab. Anim. Res. 2021, 37, 23.
- Bunag, R.D.; Tomita, T.; Sasaki, S. Streptozotocin diabetic rats are hypertensive despite reduced hypothalamic responsiveness. Hypertension 1982, 4, 556–565.
- Bagi, Z.; Erdei, N.; Toth, A.; Li, W.; Hintze, T.H.; Koller, A.; Kaley, G. Type 2 diabetic mice have increased arteriolar tone and blood pressure: Enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1610–1616.
- Senador, D.; Kanakamedala, K.; Irigoyen, M.C.; Morris, M.; Elased, K.M. Cardiovascular and autonomic phenotype of db/db diabetic mice. Exp. Physiol. 2009, 94, 648–658.
- Bhandari, U.; Kumar, V.; Khanna, N.; Panda, B.P. The effect of high-fat diet-induced obesity on cardiovascular toxicity in Wistar albino rats. Hum. Exp. Toxicol. 2011, 30, 1313–1321.
- Musial, D.C.; da Silva Junior, E.D.; da Silva, R.M.; Miranda-Ferreira, R.; Lima-Landman, M.T.; Jurkiewicz, A.; Garcia, A.G.; Jurkiewicz, N.H. Increase of angiotensin-converting enzyme activity and peripheral sympathetic dysfunction could contribute to hypertension development in streptozotocin-induced diabetic rats. Diab. Vasc. Dis. Res. 2013, 10, 498–504.
- Alameddine, A.; Fajloun, Z.; Bourreau, J.; Gauquelin-Koch, G.; Yuan, M.; Gauguier, D.; Derbre, S.; Ayer, A.; Custaud, M.A.; Navasiolava, N. The cardiovascular effects of salidroside in the Goto-Kakizaki diabetic rat model. J. Physiol. Pharmacol. 2015, 66, 249–257.
- Ma, Y.G.; Wang, J.W.; Bai, Y.G.; Liu, M.; Xie, M.J.; Dai, Z.J. Salidroside contributes to reducing blood pressure and alleviating cerebrovascular contractile activity in diabetic Goto-Kakizaki Rats by inhibition of L-type calcium channel in smooth muscle cells. BMC Pharmacol. Toxicol. 2017, 18, 30.
- Wang, Y.W.; Yu, H.R.; Tiao, M.M.; Tain, Y.L.; Lin, I.C.; Sheen, J.M.; Lin, Y.J.; Chang, K.A.; Chen, C.C.; Tsai, C.C.; et al. Maternal Obesity Related to High Fat Diet Induces Placenta Remodeling and Gut Microbiome Shaping That Are Responsible for Fetal Liver Lipid Dysmetabolism. Front. Nutr. 2021, 8, 736944.
- Carrillo-Sepulveda, M.A.; Maddie, N.; Johnson, C.M.; Burke, C.; Lutz, O.; Yakoub, B.; Kramer, B.; Persand, D. Vascular hyperacetylation is associated with vascular smooth muscle dysfunction in a rat model of non-obese type 2 diabetes. Mol. Med. 2022, 28, 30.
- Choi, S.K.; Kwon, Y.; Byeon, S.; Lee, Y.H. Stimulation of autophagy improves vascular function in the mesenteric arteries of type 2 diabetic mice. Exp. Physiol. 2020, 105, 192–200.
- Pires, P.W.; Jackson, W.F.; Dorrance, A.M. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H127–H136.
- McCurley, A.; Pires, P.W.; Bender, S.B.; Aronovitz, M.; Zhao, M.J.; Metzger, D.; Chambon, P.; Hill, M.A.; Dorrance, A.M.; Mendelsohn, M.E.; et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat. Med. 2012, 18, 1429–1433.
- Tarjus, A.; Belozertseva, E.; Louis, H.; El Moghrabi, S.; Labat, C.; Lacolley, P.; Jaisser, F.; Galmiche, G. Role of smooth muscle cell mineralocorticoid receptor in vascular tone. Pflügers Arch. 2015, 467, 1643–1650.
- Callera, G.E.; Varanda, W.A.; Bendhack, L.M. Ca2+ influx is increased in 2-kidney, 1-clip hypertensive rat aorta. Hypertension 2001, 38, 592–596.
- Pratt, P.F.; Bonnet, S.; Ludwig, L.M.; Bonnet, P.; Rusch, N.J. Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 2002, 40, 214–219.
- Bannister, J.P.; Bulley, S.; Narayanan, D.; Thomas-Gatewood, C.; Luzny, P.; Pachuau, J.; Jaggar, J.H. Transcriptional upregulation of alpha2delta-1 elevates arterial smooth muscle cell voltage-dependent Ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension 2012, 60, 1006–1015.
- Liao, J.; Zhang, Y.; Ye, F.; Zhang, L.; Chen, Y.; Zeng, F.; Shi, L. Epigenetic regulation of L-type voltage-gated Ca2+ channels in mesenteric arteries of aging hypertensive rats. Hypertens. Res. 2017, 40, 441–449.
- Matsuda, K.; Lozinskaya, I.; Cox, R.H. Augmented contributions of voltage-gated Ca2+ channels to contractile responses in spontaneously hypertensive rat mesenteric arteries. Am. J. Hypertens. 1997, 10, 1231–1239.
- Cox, R.H.; Lozinskaya, I.M. Augmented calcium currents in mesenteric artery branches of the spontaneously hypertensive rat. Hypertension 1995, 26, 1060–1064.
- Ohya, Y.; Abe, I.; Fujii, K.; Takata, Y.; Fujishima, M. Voltage-dependent Ca2+ channels in resistance arteries from spontaneously hypertensive rats. Circ. Res. 1993, 73, 1090–1099.
- Lozinskaya, I.M.; Cox, R.H. Effects of age on Ca2+ currents in small mesenteric artery myocytes from Wistar-Kyoto and spontaneously hypertensive rats. Hypertension 1997, 29, 1329–1336.
- Simard, J.M.; Li, X.; Tewari, K. Increase in functional Ca2+ channels in cerebral smooth muscle with renal hypertension. Circ. Res. 1998, 82, 1330–1337.
- DuPont, J.J.; McCurley, A.; Davel, A.P.; McCarthy, J.; Bender, S.B.; Hong, K.; Yang, Y.; Yoo, J.K.; Aronovitz, M.; Baur, W.E.; et al. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight 2016, 1, e88942.
- Takimoto, K.; Li, D.; Nerbonne, J.M.; Levitan, E.S. Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J. Mol. Cell. Cardiol. 1997, 29, 3035–3042.
- Obejero-Paz, C.A.; Lakshmanan, M.; Jones, S.W.; Scarpa, A. Effects of dexamethasone on L-type calcium currents in the A7r5 smooth muscle-derived cell line. FEBS Lett. 1993, 333, 73–77.
- Ma, Y.G.; Zhang, Y.B.; Bai, Y.G.; Dai, Z.J.; Liang, L.; Liu, M.; Xie, M.J.; Guan, H.T. Berberine alleviates the cerebrovascular contractility in streptozotocin-induced diabetic rats through modulation of intracellular Ca2+ handling in smooth muscle cells. Cardiovasc. Diabetol. 2016, 15, 63.
- Wilde, D.W.; Massey, K.D.; Walker, G.K.; Vollmer, A.; Grekin, R.J. High-fat diet elevates blood pressure and cerebrovascular muscle Ca2+ current. Hypertension 2000, 35, 832–837.
- Navedo, M.F.; Takeda, Y.; Nieves-Cintron, M.; Molkentin, J.D.; Santana, L.F. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2010, 298, C211–C220.
- Youm, J.B.; Park, K.S.; Jang, Y.J.; Leem, C.H. Effects of streptozotocin and unilateral nephrectomy on L-type Ca2+ channels and membrane capacitance in arteriolar smooth muscle cells. Pflugers Arch. 2015, 467, 1689–1697.
- Blidner, A.G.; Rabinovich, G.A. ‘Sweetening’ pregnancy: Galectins at the fetomaternal interface. Am. J. Reprod. Immunol. 2013, 69, 369–382.
- Tirado-Gonzalez, I.; Freitag, N.; Barrientos, G.; Shaikly, V.; Nagaeva, O.; Strand, M.; Kjellberg, L.; Klapp, B.F.; Mincheva-Nilsson, L.; Cohen, M.; et al. Galectin-1 influences trophoblast immune evasion and emerges as a predictive factor for the outcome of pregnancy. Mol. Hum. Reprod. 2013, 19, 43–53.
- Chang, K.; Xiao, D.; Huang, X.; Xue, Z.; Yang, S.; Longo, L.D.; Zhang, L. Chronic hypoxia inhibits sex steroid hormone-mediated attenuation of ovine uterine arterial myogenic tone in pregnancy. Hypertension 2010, 56, 750–757.
- Hu, X.Q.; Xiao, D.; Zhu, R.; Huang, X.; Yang, S.; Wilson, S.; Zhang, L. Pregnancy upregulates large-conductance Ca2+-activated K+ channel activity and attenuates myogenic tone in uterine arteries. Hypertension 2011, 58, 1132–1139.
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379.
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol. Rev. 2020, 72, 801–828.
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Montezano, A.C.; Burger, D.; Ceravolo, G.S.; Yusuf, H.; Montero, M.; Touyz, R.M. Novel Nox homologues in the vasculature: Focusing on Nox4 and Nox5. Clin. Sci. 2011, 120, 131–141.
- Kumar, K.V.; Das, U.N. Are free radicals involved in the pathobiology of human essential hypertension? Free Radic. Res. Commun. 1993, 19, 59–66.
- Russo, C.; Olivieri, O.; Girelli, D.; Faccini, G.; Zenari, M.L.; Lombardi, S.; Corrocher, R. Anti-oxidant status and lipid peroxidation in patients with essential hypertension. J. Hypertens. 1998, 16, 1267–1271.
- Koska, J.; Syrova, D.; Blazicek, P.; Marko, M.; Grna, J.D.; Kvetnansky, R.; Vigas, M. Malondialdehyde, lipofuscin and activity of antioxidant enzymes during physical exercise in patients with essential hypertension. J. Hypertens. 1999, 17, 529–535.
- Redon, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Saez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101.
- Simic, D.V.; Mimic-Oka, J.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Opacic, M.; Matic, D.; Ivanovic, B.; Simic, T. Byproducts of oxidative protein damage and antioxidant enzyme activities in plasma of patients with different degrees of essential hypertension. J. Hum. Hypertens. 2006, 20, 149–155.
- Rodrigo, R.; Prat, H.; Passalacqua, W.; Araya, J.; Bachler, J.P. Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clin. Sci. 2008, 114, 625–634.
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2014, 20, 164–182.
- Minuz, P.; Patrignani, P.; Gaino, S.; Degan, M.; Menapace, L.; Tommasoli, R.; Seta, F.; Capone, M.L.; Tacconelli, S.; Palatresi, S.; et al. Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation 2002, 106, 2800–2805.
- Will, J.C.; Ford, E.S.; Bowman, B.A. Serum vitamin C concentrations and diabetes: Findings from the Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Clin. Nutr. 1999, 70, 49–52.
- De Cristofaro, R.; Rocca, B.; Vitacolonna, E.; Falco, A.; Marchesani, P.; Ciabattoni, G.; Landolfi, R.; Patrono, C.; Davi, G. Lipid and protein oxidation contribute to a prothrombotic state in patients with type 2 diabetes mellitus. J. Thromb. Haemost. 2003, 1, 250–256.
- Palanduz, S.; Ademoglu, E.; Gokkusu, C.; Tamer, S. Plasma antioxidants and type 2 diabetes mellitus. Res. Commun. Mol. Pathol. Pharmacol. 2001, 109, 309–318.
- Komosinska-Vassev, K.; Olczyk, K.; Olczyk, P.; Winsz-Szczotka, K. Effects of metabolic control and vascular complications on indices of oxidative stress in type 2 diabetic patients. Diabetes Res. Clin. Pract. 2005, 68, 207–216.
- Soliman, G.Z. Blood lipid peroxidation (superoxide dismutase, malondialdehyde, glutathione) levels in Egyptian type 2 diabetic patients. Singapore Med. J. 2008, 49, 129–136.
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta 2012, 1822, 729–736.
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626.
- Huang, K.; Liang, Y.; Wang, K.; Wu, J.; Luo, H.; Yi, B. Influence of circulating nesfatin-1, GSH and SOD on insulin secretion in the development of T2DM. Front. Public Health 2022, 10, 882686.
- Mikhail, M.S.; Anyaegbunam, A.; Garfinkel, D.; Palan, P.R.; Basu, J.; Romney, S.L. Preeclampsia and antioxidant nutrients: Decreased plasma levels of reduced ascorbic acid, alpha-tocopherol, and beta-carotene in women with preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 150–157.
- Zusterzeel, P.L.; Mulder, T.P.; Peters, W.H.; Wiseman, S.A.; Steegers, E.A. Plasma protein carbonyls in nonpregnant, healthy pregnant and preeclamptic women. Free Radic. Res. 2000, 33, 471–476.
- Madazli, R.; Benian, A.; Aydin, S.; Uzun, H.; Tolun, N. The plasma and placental levels of malondialdehyde, glutathione and superoxide dismutase in pre-eclampsia. J. Obstet. Gynaecol. 2002, 22, 477–480.
- Aydin, S.; Benian, A.; Madazli, R.; Uludag, S.; Uzun, H.; Kaya, S. Plasma malondialdehyde, superoxide dismutase, sE-selectin, fibronectin, endothelin-1 and nitric oxide levels in women with preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 113, 21–25.
- Uzun, H.; Benian, A.; Madazli, R.; Topcuoglu, M.A.; Aydin, S.; Albayrak, M. Circulating oxidized low-density lipoprotein and paraoxonase activity in preeclampsia. Gynecol. Obstet. Investig. 2005, 60, 195–200.
- Pimentel, A.M.; Pereira, N.R.; Costa, C.A.; Mann, G.E.; Cordeiro, V.S.; de Moura, R.S.; Brunini, T.M.; Mendes-Ribeiro, A.C.; Resende, A.C. L-arginine-nitric oxide pathway and oxidative stress in plasma and platelets of patients with pre-eclampsia. Hypertens. Res. 2013, 36, 783–788.
- Kao, C.K.; Morton, J.S.; Quon, A.L.; Reyes, L.M.; Lopez-Jaramillo, P.; Davidge, S.T. Mechanism of vascular dysfunction due to circulating factors in women with pre-eclampsia. Clin. Sci. 2016, 130, 539–549.
- Babic, G.M.; Markovic, S.D.; Varjacic, M.; Djordjevic, N.Z.; Nikolic, T.; Stojic, I.; Jakovljevic, V. Estradiol decreases blood pressure in association with redox regulation in preeclampsia. Clin. Exp. Hypertens. 2018, 40, 281–286.
- Taravati, A.; Tohidi, F. Comprehensive analysis of oxidative stress markers and antioxidants status in preeclampsia. Taiwan J. Obstet. Gynecol. 2018, 57, 779–790.
- San Jose, G.; Moreno, M.U.; Olivan, S.; Beloqui, O.; Fortuno, A.; Diez, J.; Zalba, G. Functional effect of the p22phox -930A/G polymorphism on p22phox expression and NADPH oxidase activity in hypertension. Hypertension 2004, 44, 163–169.
- Wu, R.; Millette, E.; Wu, L.; de Champlain, J. Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J. Hypertens. 2001, 19, 741–748.
- Dantas, A.P.; Franco Mdo, C.; Silva-Antonialli, M.M.; Tostes, R.C.; Fortes, Z.B.; Nigro, D.; Carvalho, M.H. Gender differences in superoxide generation in microvessels of hypertensive rats: Role of NAD(P)H-oxidase. Cardiovasc. Res. 2004, 61, 22–29.
- Zalba, G.; Beaumont, F.J.; San Jose, G.; Fortuno, A.; Fortuno, M.A.; Etayo, J.C.; Diez, J. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 2000, 35, 1055–1061.
- Paravicini, T.M.; Chrissobolis, S.; Drummond, G.R.; Sobey, C.G. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke 2004, 35, 584–589.
- Lodi, F.; Cogolludo, A.; Duarte, J.; Moreno, L.; Coviello, A.; Peral De Bruno, M.; Vera, R.; Galisteo, M.; Jimenez, R.; Tamargo, J.; et al. Increased NADPH oxidase activity mediates spontaneous aortic tone in genetically hypertensive rats. Eur. J. Pharmacol. 2006, 544, 97–103.
- Akasaki, T.; Ohya, Y.; Kuroda, J.; Eto, K.; Abe, I.; Sumimoto, H.; Iida, M. Increased expression of gp91phox homologues of NAD(P)H oxidase in the aortic media during chronic hypertension: Involvement of the renin-angiotensin system. Hypertens. Res. 2006, 29, 813–820.
- Wind, S.; Beuerlein, K.; Armitage, M.E.; Taye, A.; Kumar, A.H.; Janowitz, D.; Neff, C.; Shah, A.M.; Wingler, K.; Schmidt, H.H. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension 2010, 56, 490–497.
- Camargo, L.L.; Harvey, A.P.; Rios, F.J.; Tsiropoulou, S.; Da Silva, R.N.O.; Cao, Z.; Graham, D.; McMaster, C.; Burchmore, R.J.; Hartley, R.C.; et al. Vascular Nox (NADPH Oxidase) Compartmentalization, Protein Hyperoxidation, and Endoplasmic Reticulum Stress Response in Hypertension. Hypertension 2018, 72, 235–246.
- Beswick, R.A.; Dorrance, A.M.; Leite, R.; Webb, R.C. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001, 38, 1107–1111.
- Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Yogi, A.; He, Y.; Malkinson, S.; Schiffrin, E.L. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 2005, 45, 773–779.
- Miyata, K.; Rahman, M.; Shokoji, T.; Nagai, Y.; Zhang, G.X.; Sun, G.P.; Kimura, S.; Yukimura, T.; Kiyomoto, H.; Kohno, M.; et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J. Am. Soc. Nephrol. 2005, 16, 2906–2912.
- Chrissobolis, S.; Drummond, G.R.; Faraci, F.M.; Sobey, C.G. Chronic aldosterone administration causes Nox2-mediated increases in reactive oxygen species production and endothelial dysfunction in the cerebral circulation. J. Hypertens. 2014, 32, 1815–1821.
- Siuda, D.; Tobias, S.; Rus, A.; Xia, N.; Forstermann, U.; Li, H. Dexamethasone upregulates Nox1 expression in vascular smooth muscle cells. Pharmacology 2014, 94, 13–20.
- Gayen, J.R.; Zhang, K.; RamachandraRao, S.P.; Mahata, M.; Chen, Y.; Kim, H.S.; Naviaux, R.K.; Sharma, K.; Mahata, S.K.; O’Connor, D.T. Role of reactive oxygen species in hyperadrenergic hypertension: Biochemical, physiological, and pharmacological evidence from targeted ablation of the chromogranin a (Chga) gene. Circ. Cardiovasc. Genet. 2010, 3, 414–425.
- Cifuentes, M.E.; Rey, F.E.; Carretero, O.A.; Pagano, P.J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2234–H2240.
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676.
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q.t.; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51.
- Mollnau, H.; Wendt, M.; Szocs, K.; Lassegue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, E58–E65.
- Virdis, A.; Neves, M.F.; Amiri, F.; Touyz, R.M.; Schiffrin, E.L. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J. Hypertens. 2004, 22, 535–542.
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685.
- Wang, D.; Chabrashvili, T.; Borrego, L.; Aslam, S.; Umans, J.G. Angiotensin II infusion alters vascular function in mouse resistance vessels: Roles of O and endothelium. J. Vasc. Res. 2006, 43, 109–119.
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684.
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213.
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662.
- Xi, G.; Shen, X.; Maile, L.A.; Wai, C.; Gollahon, K.; Clemmons, D.R. Hyperglycemia enhances IGF-I-stimulated Src activation via increasing Nox4-derived reactive oxygen species in a PKCzeta-dependent manner in vascular smooth muscle cells. Diabetes 2012, 61, 104–113.
- Lu, T.; Chai, Q.; Yu, L.; d’Uscio, L.V.; Katusic, Z.S.; He, T.; Lee, H.C. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel beta1 subunit degradation in diabetic mice. Diabetes 2012, 61, 1860–1868.
- Wendt, M.C.; Daiber, A.; Kleschyov, A.L.; Mulsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J.F., Jr.; Lassegue, B.; Walter, U.; et al. Differential effects of diabetes on the expression of the gp91phox homologues nox1 and nox4. Free Radic. Biol. Med. 2005, 39, 381–391.
- Ding, H.; Hashem, M.; Triggle, C. Increased oxidative stress in the streptozotocin-induced diabetic apoE-deficient mouse: Changes in expression of NADPH oxidase subunits and eNOS. Eur. J. Pharmacol. 2007, 561, 121–128.
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626.
- Rezende, F.; Moll, F.; Walter, M.; Helfinger, V.; Hahner, F.; Janetzko, P.; Ringel, C.; Weigert, A.; Fleming, I.; Weissmann, N.; et al. The NADPH organizers NoxO1 and p47phox are both mediators of diabetes-induced vascular dysfunction in mice. Redox Biol. 2018, 15, 12–21.
- Manea, S.A.; Antonescu, M.L.; Fenyo, I.M.; Raicu, M.; Simionescu, M.; Manea, A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 2018, 16, 332–343.
- San Martin, A.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Gongora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2073–H2082.
- Kassan, M.; Choi, S.K.; Galan, M.; Lee, Y.H.; Trebak, M.; Matrougui, K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 MAP kinase-dependent mechanisms in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H972–H980.
- Bruder-Nascimento, T.; Callera, G.E.; Montezano, A.C.; He, Y.; Antunes, T.T.; Nguyen Dinh Cat, A.; Tostes, R.C.; Touyz, R.M. Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: Role of Rac1/2-sensitive Nox-dependent pathways. Clin. Sci. 2015, 128, 411–423.
- Liu, S.; Ma, X.; Gong, M.; Shi, L.; Lincoln, T.; Wang, S. Glucose down-regulation of cGMP-dependent protein kinase I expression in vascular smooth muscle cells involves NAD(P)H oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2007, 42, 852–863.
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell. Physiol. 2007, 212, 682–689.
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes 2003, 52, 2795–2804.
- Weidig, P.; McMaster, D.; Bayraktutan, U. High glucose mediates pro-oxidant and antioxidant enzyme activities in coronary endothelial cells. Diabetes Obes. Metab. 2004, 6, 432–441.
- Ulker, S.; McMaster, D.; McKeown, P.P.; Bayraktutan, U. Antioxidant vitamins C and E ameliorate hyperglycaemia-induced oxidative stress in coronary endothelial cells. Diabetes Obes. Metab. 2004, 6, 442–451.
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904.
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541.
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.t.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452.
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell. Mol. Biol. 2005, 51, 573–578.
- Rodriguez-Iturbe, B.; Sepassi, L.; Quiroz, Y.; Ni, Z.; Wallace, D.C.; Vaziri, N.D. Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. J. Appl. Physiol. 2007, 102, 255–260.
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574.
- Porter, G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176.
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227.
- Viel, E.C.; Benkirane, K.; Javeshghani, D.; Touyz, R.M.; Schiffrin, E.L. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H281–H288.
- Peng, S.Y.; Tsai, C.H.; Wu, X.M.; Huang, H.H.; Chen, Z.W.; Lee, B.C.; Chang, Y.Y.; Pan, C.T.; Wu, V.C.; Chou, C.H.; et al. Aldosterone Suppresses Endothelial Mitochondria through Mineralocorticoid Receptor/Mitochondrial Reactive Oxygen Species Pathway. Biomedicines 2022, 10, 1119.
- Kizhakekuttu, T.J.; Wang, J.; Dharmashankar, K.; Ying, R.; Gutterman, D.D.; Vita, J.A.; Widlansky, M.E. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2531–2539.
- Mackenzie, R.M.; Salt, I.P.; Miller, W.H.; Logan, A.; Ibrahim, H.A.; Degasperi, A.; Dymott, J.A.; Hamilton, C.A.; Murphy, M.P.; Delles, C.; et al. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin. Sci. 2013, 124, 403–411.
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794.
- Cho, Y.E.; Basu, A.; Dai, A.; Heldak, M.; Makino, A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am. J. Physiol. Cell. Physiol. 2013, 305, C1033–C1040.
- Keller, A.C.; Knaub, L.A.; McClatchey, P.M.; Connon, C.A.; Bouchard, R.; Miller, M.W.; Geary, K.E.; Walker, L.A.; Klemm, D.J.; Reusch, J.E. Differential Mitochondrial Adaptation in Primary Vascular Smooth Muscle Cells from a Diabetic Rat Model. Oxid. Med. Cell. Longev. 2016, 2016, 8524267.
- Merdzo, I.; Rutkai, I.; Sure, V.N.; McNulty, C.A.; Katakam, P.V.; Busija, D.W. Impaired Mitochondrial Respiration in Large Cerebral Arteries of Rats with Type 2 Diabetes. J. Vasc. Res. 2017, 54, 1–12.
- Merdzo, I.; Rutkai, I.; Sure, V.; Katakam, P.V.G.; Busija, D.W. Effects of prolonged type 2 diabetes on mitochondrial function in cerebral blood vessels. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1086–H1092.
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790.
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Zuodar, G.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis 2005, 183, 259–267.
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351.
- Xie, L.; Zhu, X.; Hu, Y.; Li, T.; Gao, Y.; Shi, Y.; Tang, S. Mitochondrial DNA oxidative damage triggering mitochondrial dysfunction and apoptosis in high glucose-induced HRECs. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4203–4209.
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1876–H1881.
- Sun, J.; Xu, Y.; Sun, S.; Sun, Y.; Wang, X. Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: The role of mitochondrial reactive oxygen species. Mol. Cell. Biochem. 2010, 343, 27–35.
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453.
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52.
- Yoshinaga, A.; Kajihara, N.; Kukidome, D.; Motoshima, H.; Matsumura, T.; Nishikawa, T.; Araki, E. Hypoglycemia Induces Mitochondrial Reactive Oxygen Species Production Through Increased Fatty Acid Oxidation and Promotes Retinal Vascular Permeability in Diabetic Mice. Antioxid. Redox Signal. 2021, 34, 1245–1259.
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829.
- Togliatto, G.; Lombardo, G.; Brizzi, M.F. The Future Challenge of Reactive Oxygen Species (ROS) in Hypertension: From Bench to Bed Side. Int. J. Mol. Sci. 2017, 18, 1988.
- Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Vazquez, C.M.; Mate, A.; Sobrevia, L.; Marin, R. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165354.
- Hu, X.Q.; Zhang, L. Hypoxia and Mitochondrial Dysfunction in Pregnancy Complications. Antioxidants 2021, 10, 405.
- Hu, X.Q.; Zhang, L. Mitochondrial Dysfunction in the Pathogenesis of Preeclampsia. Curr. Hypertens. Rep. 2022, 24, 157–172.
- Schnackenberg, C.G.; Wilcox, C.S. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2alpha. Hypertension 1999, 33, 424–428.
- Welch, W.J.; Mendonca, M.; Aslam, S.; Wilcox, C.S. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension 2003, 41, 692–696.
- Beswick, R.A.; Zhang, H.; Marable, D.; Catravas, J.D.; Hill, W.D.; Webb, R.C. Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension 2001, 37, 781–786.
- Schnackenberg, C.G.; Welch, W.J.; Wilcox, C.S. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: Role of nitric oxide. Hypertension 1998, 32, 59–64.
- Park, J.B.; Touyz, R.M.; Chen, X.; Schiffrin, E.L. Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 2002, 15, 78–84.
- Palm, F.; Onozato, M.; Welch, W.J.; Wilcox, C.S. Blood pressure, blood flow, and oxygenation in the clipped kidney of chronic 2-kidney, 1-clip rats: Effects of tempol and Angiotensin blockade. Hypertension 2010, 55, 298–304.
- Brands, M.W.; Bell, T.D.; Gibson, B. Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension 2004, 43, 57–63.
- Onuma, S.; Nakanishi, K. Superoxide dismustase mimetic tempol decreases blood pressure by increasing renal medullary blood flow in hyperinsulinemic-hypertensive rats. Metabolism 2004, 53, 1305–1308.
- Peixoto, E.B.; Pessoa, B.S.; Biswas, S.K.; Lopes de Faria, J.B. Antioxidant SOD mimetic prevents NADPH oxidase-induced oxidative stress and renal damage in the early stage of experimental diabetes and hypertension. Am. J. Nephrol. 2009, 29, 309–318.
- Li, H.; Liu, X.; Ren, Z.; Gu, J.; Lu, Y.; Wang, X.; Zhang, L. Effects of Diabetic Hyperglycemia on Central Ang-(1-7)-Mas-R-nNOS Pathways in Spontaneously Hypertensive Rats. Cell. Physiol. Biochem. 2016, 40, 1186–1197.
- Sedeek, M.; Gilbert, J.S.; LaMarca, B.B.; Sholook, M.; Chandler, D.L.; Wang, Y.; Granger, J.P. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am. J. Hypertens. 2008, 21, 1152–1156.
- Parrish, M.R.; Wallace, K.; Tam Tam, K.B.; Herse, F.; Weimer, A.; Wenzel, K.; Wallukat, G.; Ray, L.F.; Arany, M.; Cockrell, K.; et al. Hypertension in response to AT1-AA: Role of reactive oxygen species in pregnancy-induced hypertension. Am. J. Hypertens. 2011, 24, 835–840.
- Tam Tam, K.B.; Lamarca, B.; Arany, M.; Cockrell, K.; Fournier, L.; Murphy, S.; Martin, J.N., Jr.; Granger, J.P. Role of reactive oxygen species during hypertension in response to chronic antiangiogenic factor (sFlt-1) excess in pregnant rats. Am. J. Hypertens. 2011, 24, 110–113.
- Hoffmann, D.S.; Weydert, C.J.; Lazartigues, E.; Kutschke, W.J.; Kienzle, M.F.; Leach, J.E.; Sharma, J.A.; Sharma, R.V.; Davisson, R.L. Chronic tempol prevents hypertension, proteinuria, and poor feto-placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 2008, 51, 1058–1065.
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593.
- Carlstrom, M.; Brown, R.D.; Sallstrom, J.; Larsson, E.; Zilmer, M.; Zabihi, S.; Eriksson, U.J.; Persson, A.E. SOD1 deficiency causes salt sensitivity and aggravates hypertension in hydronephrosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R82–R92.
- Zhou, X.; Bohlen, H.G.; Miller, S.J.; Unthank, J.L. NAD(P)H oxidase-derived peroxide mediates elevated basal and impaired flow-induced NO production in SHR mesenteric arteries in vivo. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1008–H1016.
- Lim, R.; Acharya, R.; Delpachitra, P.; Hobson, S.; Sobey, C.G.; Drummond, G.R.; Wallace, E.M. Activin and NADPH-oxidase in preeclampsia: Insights from in vitro and murine studies. Am. J. Obstet. Gynecol. 2015, 212, 86.e1–86.e12.
- Baumer, A.T.; Kruger, C.A.; Falkenberg, J.; Freyhaus, H.T.; Rosen, R.; Fink, K.; Rosenkranz, S. The NAD(P)H oxidase inhibitor apocynin improves endothelial NO/superoxide balance and lowers effectively blood pressure in spontaneously hypertensive rats: Comparison to calcium channel blockade. Clin. Exp. Hypertens. 2007, 29, 287–299.
- Unger, B.S.; Patil, B.M. Apocynin improves endothelial function and prevents the development of hypertension in fructose fed rat. Indian J. Pharmacol. 2009, 41, 208–212.
- Guimaraes, D.D.; Carvalho, C.C.; Braga, V.A. Scavenging of NADPH oxidase-derived superoxide anions improves depressed baroreflex sensitivity in spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2012, 39, 373–378.
- Wallace, K.; Cornelius, D.C.; Scott, J.; Heath, J.; Moseley, J.; Chatman, K.; LaMarca, B. CD4+ T cells are important mediators of oxidative stress that cause hypertension in response to placental ischemia. Hypertension 2014, 64, 1151–1158.
- Perassa, L.A.; Graton, M.E.; Potje, S.R.; Troiano, J.A.; Lima, M.S.; Vale, G.T.; Pereira, A.A.; Nakamune, A.C.; Sumida, D.H.; Tirapelli, C.R.; et al. Apocynin reduces blood pressure and restores the proper function of vascular endothelium in SHR. Vasc. Pharmacol. 2016, 87, 38–48.
- Dikalova, A.E.; Gongora, M.C.; Harrison, D.G.; Lambeth, J.D.; Dikalov, S.; Griendling, K.K. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H673–H679.
- Hool, L.C. The L-type Ca2+ channel as a potential mediator of pathology during alterations in cellular redox state. Heart Lung Circ. 2009, 18, 3–10.
- Scragg, J.L.; Dallas, M.L.; Wilkinson, J.A.; Varadi, G.; Peers, C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J. Biol. Chem. 2008, 283, 24412–24419.
- Muralidharan, P.; Cserne Szappanos, H.; Ingley, E.; Hool, L. Evidence for redox sensing by a human cardiac calcium channel. Sci. Rep. 2016, 6, 19067.
- Amberg, G.C.; Earley, S.; Glapa, S.A. Local regulation of arterial L-type calcium channels by reactive oxygen species. Circ. Res. 2010, 107, 1002–1010.
- Chaplin, N.L.; Amberg, G.C. Hydrogen peroxide mediates oxidant-dependent stimulation of arterial smooth muscle L-type calcium channels. Am. J. Physiol. Cell. Physiol. 2012, 302, C1382–C1393.
- Sotnikova, R. Investigation of the mechanisms underlying H2O2-evoked contraction in the isolated rat aorta. Gen. Pharmacol. 1998, 31, 115–119.
- Yang, Z.W.; Zheng, T.; Wang, J.; Zhang, A.; Altura, B.T.; Altura, B.M. Hydrogen peroxide induces contraction and raises i in canine cerebral arterial smooth muscle: Participation of cellular signaling pathways. Naunyn-Schmiedeberg Arch. Pharmacol. 1999, 360, 646–653.
- Tabet, F.; Savoia, C.; Schiffrin, E.L.; Touyz, R.M. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 2004, 44, 200–208.
- Garcia-Redondo, A.B.; Briones, A.M.; Beltran, A.E.; Alonso, M.J.; Simonsen, U.; Salaices, M. Hypertension increases contractile responses to hydrogen peroxide in resistance arteries through increased thromboxane A2, Ca2+, and superoxide anion levels. J. Pharmacol. Exp. Ther. 2009, 328, 19–27.
- Garcia-Redondo, A.B.; Briones, A.M.; Avendano, M.S.; Hernanz, R.; Alonso, M.J.; Salaices, M. Losartan and tempol treatments normalize the increased response to hydrogen peroxide in resistance arteries from hypertensive rats. J. Hypertens. 2009, 27, 1814–1822.
- Santiago, E.; Contreras, C.; Garcia-Sacristan, A.; Sanchez, A.; Rivera, L.; Climent, B.; Prieto, D. Signaling pathways involved in the H2O2-induced vasoconstriction of rat coronary arteries. Free Radic. Biol. Med. 2013, 60, 136–146.
- Garcia-Redondo, A.B.; Briones, A.M.; Martinez-Revelles, S.; Palao, T.; Vila, L.; Alonso, M.J.; Salaices, M. c-Src, ERK1/2 and Rho kinase mediate hydrogen peroxide-induced vascular contraction in hypertension: Role of TXA2, NAD(P)H oxidase and mitochondria. J. Hypertens. 2015, 33, 77–87.
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404.
- Hu, Z.; Zhang, B.; Lim, L.J.Y.; Loh, W.Z.K.; Yu, D.; Tan, B.W.Q.; Liang, M.C.; Huang, Z.; Leo, C.H.; Huang, H.; et al. S-Nitrosylation-Mediated Reduction of Cav1.2 Surface Expression and Open Probability Underlies Attenuated Vasoconstriction Induced by Nitric Oxide. Hypertension 2022, 79, 2854–2866.
- Poteser, M.; Romanin, C.; Schreibmayer, W.; Mayer, B.; Groschner, K. S-nitrosation controls gating and conductance of the alpha 1 subunit of class C L-type Ca2+ channels. J. Biol. Chem. 2001, 276, 14797–14803.
- Schulz, E.; Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673.
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101.
- Liu, H.; Colavitti, R.; Rovira, I.I.; Finkel, T. Redox-dependent transcriptional regulation. Circ. Res. 2005, 97, 967–974.
- Wang, W.Z.; Pang, L.; Palade, P. Angiotensin II causes endothelial-dependent increase in expression of Cav1.2 protein in cultured arteries. Eur. J. Pharmacol. 2008, 599, 117–120.
- Wang, W.; Pang, L.; Palade, P. Angiotensin II upregulates Cav1.2 protein expression in cultured arteries via endothelial H2O2 production. J. Vasc. Res. 2011, 48, 67–78.
- Tsai, C.T.; Wang, D.L.; Chen, W.P.; Hwang, J.J.; Hsieh, C.S.; Hsu, K.L.; Tseng, C.D.; Lai, L.P.; Tseng, Y.Z.; Chiang, F.T.; et al. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element-binding protein-dependent pathway in HL-1 myocytes. Circ. Res. 2007, 100, 1476–1485.
- Lerman, L.O.; Nath, K.A.; Rodriguez-Porcel, M.; Krier, J.D.; Schwartz, R.S.; Napoli, C.; Romero, J.C. Increased oxidative stress in experimental renovascular hypertension. Hypertension 2001, 37, 541–546.
- Cervenka, L.; Horacek, V.; Vaneckova, I.; Hubacek, J.A.; Oliverio, M.I.; Coffman, T.M.; Navar, L.G. Essential role of AT1A receptor in the development of 2K1C hypertension. Hypertension 2002, 40, 735–741.
- Cervenka, L.; Vaneckova, I.; Huskova, Z.; Vanourkova, Z.; Erbanova, M.; Thumova, M.; Skaroupkova, P.; Opocensky, M.; Maly, J.; Chabova, V.C.; et al. Pivotal role of angiotensin II receptor subtype 1A in the development of two-kidney, one-clip hypertension: Study in angiotensin II receptor subtype 1A knockout mice. J. Hypertens. 2008, 26, 1379–1389.
- Isidori, A.M.; Graziadio, C.; Paragliola, R.M.; Cozzolino, A.; Ambrogio, A.G.; Colao, A.; Corsello, S.M.; Pivonello, R.; Group, A.B.C.S. The hypertension of Cushing’s syndrome: Controversies in the pathophysiology and focus on cardiovascular complications. J. Hypertens. 2015, 33, 44–60.
- Barbot, M.; Ceccato, F.; Scaroni, C. The Pathophysiology and Treatment of Hypertension in Patients With Cushing’s Syndrome. Front. Endocrinol. 2019, 10, 321.
- Schewe, J.; Seidel, E.; Forslund, S.; Marko, L.; Peters, J.; Muller, D.N.; Fahlke, C.; Stolting, G.; Scholl, U. Elevated aldosterone and blood pressure in a mouse model of familial hyperaldosteronism with ClC-2 mutation. Nat. Commun. 2019, 10, 5155.
- Nowicki, P.T.; Flavahan, S.; Hassanain, H.; Mitra, S.; Holland, S.; Goldschmidt-Clermont, P.J.; Flavahan, N.A. Redox signaling of the arteriolar myogenic response. Circ. Res. 2001, 89, 114–116.
- Cseko, C.; Bagi, Z.; Koller, A. Biphasic effect of hydrogen peroxide on skeletal muscle arteriolar tone via activation of endothelial and smooth muscle signaling pathways. J. Appl. Physiol. 2004, 97, 1130–1137.
- Li, L.; Lai, E.Y.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Differential effects of superoxide and hydrogen peroxide on myogenic signaling, membrane potential, and contractions of mouse renal afferent arterioles. Am. J. Physiol. Renal. Physiol. 2016, 310, F1197–F1205.
- Wagenfeld, L.; von Domarus, F.; Weiss, S.; Klemm, M.; Richard, G.; Zeitz, O. The effect of reactive oxygen species on the myogenic tone of rat ophthalmic arteries with and without endothelium. Graefe Arch. Clin. Exp. Ophthalmol. 2013, 251, 2339–2344.
- Spurrell, B.E.; Murphy, T.V.; Hill, M.A. Tyrosine phosphorylation modulates arteriolar tone but is not fundamental to myogenic response. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H373–H382.
- Murphy, T.V.; Spurrell, B.E.; Hill, M.A. Tyrosine phosphorylation following alterations in arteriolar intraluminal pressure and wall tension. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1047–H1056.
- Xiao, D.; Buchholz, J.N.; Zhang, L. Pregnancy attenuates uterine artery pressure-dependent vascular tone: Role of PKC/ERK pathway. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2337–H2343.
- Osol, G.; Laher, I.; Cipolla, M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ. Res. 1991, 68, 359–367.
- Hill, M.A.; Falcone, J.C.; Meininger, G.A. Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am. J. Physiol. 1990, 259, H1586–H1594.
- Chang, K.; Xiao, D.; Huang, X.; Longo, L.D.; Zhang, L. Chronic hypoxia increases pressure-dependent myogenic tone of the uterine artery in pregnant sheep: Role of ERK/PKC pathway. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1840–H1849.
- Kizub, I.V.; Pavlova, O.O.; Johnson, C.D.; Soloviev, A.I.; Zholos, A.V. Rho kinase and protein kinase C involvement in vascular smooth muscle myofilament calcium sensitization in arteries from diabetic rats. Br. J. Pharmacol. 2010, 159, 1724–1731.
- Novokhatska, T.; Tishkin, S.; Dosenko, V.; Boldyriev, A.; Ivanova, I.; Strielkov, I.; Soloviev, A. Correction of vascular hypercontractility in spontaneously hypertensive rats using shRNAs-induced delta protein kinase C gene silencing. Eur. J. Pharmacol. 2013, 718, 401–407.
- Ungvari, Z.; Pacher, P.; Kecskemeti, V.; Papp, G.; Szollar, L.; Koller, A. Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc. Res. 1999, 43, 1018–1028.
- Shaikh, A. A Practical Approach to Hypertension Management in Diabetes. Diabetes Ther. 2017, 8, 981–989.
- Tocci, G.; Desideri, G.; Roca, E.; Calcullo, C.; Crippa, M.; De Luca, N.; Gaudio, G.V.; Lonati, L.M.; Orselli, L.; Scuteri, A.; et al. How to Improve Effectiveness and Adherence to Antihypertensive Drug Therapy: Central Role of Dihydropyridinic Calcium Channel Blockers in Hypertension. High Blood Press. Cardiovasc. Prev. 2018, 25, 25–34.
- Odigboegwu, O.; Pan, L.J.; Chatterjee, P. Use of Antihypertensive Drugs During Preeclampsia. Front. Cardiovasc. Med. 2018, 5, 50.
- Ojha, U.; Ruddaraju, S.; Sabapathy, N.; Ravindran, V.; Worapongsatitaya, P.; Haq, J.; Mohammed, R.; Patel, V. Current and Emerging Classes of Pharmacological Agents for the Management of Hypertension. Am. J. Cardiovasc. Drugs 2022, 22, 271–285.