Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jong-Wha Jung and Version 2 by Jessie Wu.
Autophagy is a cellular process that removes damaged components of cells and recycles them as biochemical building blocks. Autophagy can also be induced to protect cells in response to intra- and extracellular stresses, including damage to cellular components, nutrient deprivation, hypoxia, and pathogenic invasion. Dysregulation of autophagy has been attributed to various diseases. In particular, autophagy protects cancer cells by supporting tumor cell survival and the development of drug resistance. The ULK complex is an early-stage regulator of autophagy and attracted particular attention as a drug target. Among ULK isoforms, ULK1, ULK2, ULK3, ULK4, and serine/threonine-protein kinase 36 (STK36), ULK1 have been most extensively studied.
- Unc-51-like autophagy-activating kinase
- autophagy
- cancer
1. Discovery and Development of Unc-51-like Autophagy-Activating LKinase Inhibitors
Functional, physiological, and structural studies on Unc-51-like autophagy-activating kinases (ULKs) in autophagy processes have led to the discovery and development of ULK inhibitors. The structures of these small-molecule inhibitors are shown in Figure 1. Target-based screening of kinase-related chemical libraries in a reverse pharmacology approach was a popular method to discover ULK inhibitors possessing novel scaffolds of 2-aminopyrimidine, pyrazole, or indole. A chemical library was derived from a focal adhesion kinase (FAK) inhibitor exhibiting cross-reactivity toward ULK1, and in vitro ULK1 kinase screening of the library led to the discovery of a 2-aminopyrimidine compound, SBI-0206965 [1][38]. Although SBI-0206965 is a potent ULK1 inhibitor, its poor absorption after oral administration and poor systemic exposure following intraperitoneal dosing in mice has limited its use in vivo. To improve the potency and drug-like properties of SBI-0206965, a structure-based rational design was used to generate SBP-7455 [2][39]. Screening with a P-ATP radioactive assay revealed pyrazole aminoquinoline compounds as potent ULK1 inhibitors [3][21]. Remarkably, a selected pyrazole aminoquinoline derivative dramatically stabilized the ULK1 KD, which aided the crystallization of ULK1. Using the ligand-bound crystal structure, compound 6 possessing pyrazole aminoquinoline scaffold was developed as the most potent ULK1 inhibitor among the series. Although compound 6 had high affinity in inhibition of ULK1, it’s activity was not specific to ULK1, and it was not suitable for cellular use. Thus, screening continued for another lead compound, and compound 3 was identified as a potent and selective ULK1 inhibitor for further development [4][40]. MRT67307 and MRT68921, initially synthesized as TBK1 inhibitors possessing 2-aminopyrimidine scaffold, were also discovered as potent inhibitors for both ULK1 and ULK2. Screening of a kinase-focused library using differential scanning fluorimetry revealed Aurora A kinase inhibitors, such as PF-03814735 and hesperidin, as potent binders for both ULK1 and ULK2 [5][22]. ULK1 activity screening led to the discovery of pyrazolopyrimidine compounds including ULK-101 [6][18]. GSK Published Kinase Inhibitor Sets were screened for ULK inhibition, and GW837331X and GW406108X were identified as potent ULK1 inhibitors [7][41].
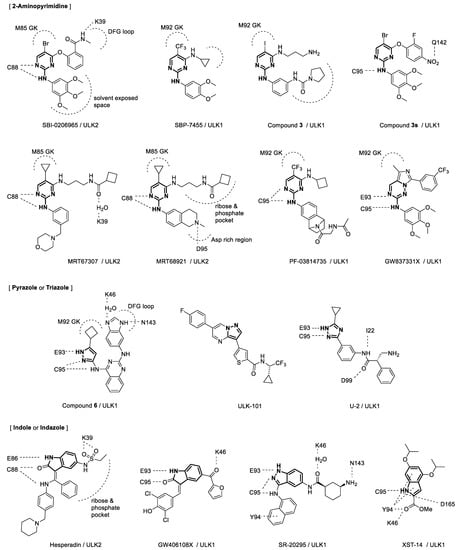
Figure 1.
Structures of ULK1/2 inhibitors and key interactions with their binding sites. GK = gate keeper.
Virtual screening methods also have been employed for the discovery of ULK inhibitors. Docking-based pharmacophore modeling led to the discovery of 2-aminopyrimidine compound 3s, as a potent ULK1 inhibitor [8][42]. In silico high-throughput screening, combined with a biochemical assay, identified an indazole compound as an ULK inhibitor [9][43]. Further optimization led to the development of SR-20295 with nanomolar potency. Structure-based virtual docking with visual inspection and in silico filtering for drug-like properties highlighted an indole compound as a potent ULK1 inhibitor including XST-14 [10][44]. Pharmacophore-based virtual screening combined with structure-based docking led to the development of U-2, which exhibited a sub-micromolar IC50 value against ULK1 [11][45].
2. Biological and Anticancer Effects of Unc-51-like Autophagy-Activating LKinase Inhibitors
Biological effects of ULK inhibitors including anticancer effects against specific cell lines are summarized in Table 1. SBI-0206965 is one of the most extensively explored ULK1 inhibitors and a potential anticancer agent [1][38]. SBI-0206965 is a potent inhibitor for both ULK1 and ULK2. SBI-0206965 suppresses mTOR kinase inhibitor-induced ULK1-dependent autophagy in A549 lung cancer cells. It also promotes apoptosis via inducing the loss of autophagic maintenance involved in cancer cell survival [1][38]. Furthermore, SBI-0206965 suppresses cell growth and promotes apoptosis in neuroblastoma cell lines [12][46]. SBI-0206965 triggers apoptosis in clear cell renal cell carcinoma cells by upregulating ULK1 mRNA expression and inhibiting xenograft tumor growth in mice [13][47]. SBI-0206965 induces caspase-dependent apoptosis in FLT3-ITD-mutated acute myeloid leukemia (AML) cells by inhibiting autophagy and increasing the reactive oxygen species levels [14][25]. Although SBI-0206965 is a potent ULK1 inhibitor, its poor absorption after oral administration and poor systemic exposure following intraperitoneal dosing in mice has limited its use in vivo [2][39].
SBP-7455 exhibited improved potency and binding affinity for both ULK1 and ULK2 compared with those of SBI-0206965 [2][39]. SBP-7455 significantly reduced ULK1-dependent Beclin1 and VPS34 phosphorylation, and inhibited autophagy in cells. Importantly, SBP-7455 demonstrated improved drug-like properties, such as improved physicochemical and pharmacokinetic profiles. After per oral administration of SBP-7455, downregulation of total ULK1 and ATG13 levels, as well as inhibition of ATG13 phosphorylation by ULK1, were observed in mouse liver samples [2][39]. Notably, SBP-7455 effectively reduced the viability of triple-negative breast cancer (TNBC) cells [2][39]. Furthermore, it induced high cytotoxicity in TNBC cells and increased apoptosis under starvation conditions. Moreover, it inhibited autophagic flux and decreased the number of autophagosomes.
Compound 6 is a highly potent ULK1 inhibitor with a single digit nanomolar IC50, but it was not suitable for cellular use because its activity was not specific to ULK1 [3][21]. Compound 3 is not as potent as compound 6, but it is a potent and selective ULK1 inhibitor which enabled further development in cells [4][40]. Compound 3 inhibited autophagy in cells via ULK1, as evidenced by the accumulation of LC3-I relative to that of LC3-II, a common marker of autophagosome formation.
MRT68921 exhibited nanomolar IC50 values for both ULK1 and ULK2 [15][24]. It also suppressed autophagy in MEF cells in a ULK1-dependent manner. MRT68921 exerts cytotoxic effects in various cancer cell lines and has a comparatively high safety window [16][48]. MRT68921 induced caspase-dependent apoptosis in FLT3-ITD-mutated AML cells by inhibiting autophagy and increasing reactive oxygen species, similar to SBI-0206965 [14][25]. Furthermore, MRT68921 diminished the c-AMP-mediated protective effect on the survival of DNA damage-induced B cell precursor acute lymphoblastic leukemia cells [17][49]. It also suppressed autophagy and viability by inhibiting ULK1 in high-grade serous ovarian cancer spheroids [18][50].
Compound 3s exhibited stronger ULK1 inhibitory activity at 10 µM than that of SBI-0206965 [8][42]. On treatment of 3s, autophagy substrate p62 increased while conversion of LC3 I to LC3II reduced together with reduction in Beclin1. i.e., compound 3s effectively blocked autophagy by inhibiting ULK1. The anti-proliferative activity of compound 3s was the most prominent in A549 lung cancer cells among five different cell lines tested, including leukemia and breast cancer cells.
PF-03814735 and hesperidin exhibited nanomolar KD values for both ULK1 and ULK2 in isothermal calorimetry experiments [5][22]. The anticancer effects of PF-03814735 and hesperidin were reported before they were identified as ULK inhibitors, and the relationship between their anticancer effects and ULK inhibitory activities remains unclear. PF-03814735 was originally developed as an Aurora A kinase A/B inhibitor and has completed a phase I clinical trial for advanced solid tumors [19][20][51,52]. Hesperidin is a known natural product with anticancer activity [21][53].
SR-20295 is a potent ULK1 inhibitor with an excellent stability against drug metabolism [9][43]. It was quite stable in human, rat, and mouse microsomes, and exhibited negligible CYP inhibition. However, further studies including its effects against ULK dependent cancers has not been disclosed yet.
ULK-101 exhibited nanomolar inhibitory activity for both ULK1 and ULK2 [6][18]. ULK-101 suppressed mTOR inhibitor-induced formation of early autophagic vesicles, omegasomes, and phagophores. ULK-101 also reduced both basal and induced autophagy. Moreover, ULK-101 reduced the survival of osteosarcoma cells in starvation media compared to that in full growth media. A KRAS mutant or amplified non-small cell lung cancer (NSCLC) cells also showed increased ULK-101 sensitivity in starvation medium. Thus, ULK-101 is suggested to suppress autophagy-dependent cancer cell survival.
XST-14 exhibited nanomolar IC50 for ULK1 [10][44]. XST-14 also inhibited ULK2, almost as effectively as ULK1. XST-14 strongly inhibited the conversion of LC3-I to LC3-II and blocked autophagosome/lysosome fusion in CHO cells, indicating a blockade of the autophagic flux. XST-14 decreased the number of LC3 puncta and autophagic vacuole volume density in starved HepG2 cells. XST-14 also inhibited the phosphorylation of downstream targets of ULK1, PIK3C3, and Beclin1, decreased their interaction with ULK1, and destabilized the PIK3C3 and Beclin1 complex in HepG2 cells subjected to starvation-induced autophagy. XST-14 decreased the proliferation and invasion of hepatocellular carcinoma cells and induced apoptosis in a ULK1-dependent manner.
GW837331X and GW406108X exhibited sub-micromolar IC50s for ULK1 [7][41]. Enzyme kinetic studies revealed that the inhibitory effects of the compounds were primarily attributed to competitive inhibition of ATP to ULK1. Both compounds also inhibited ATG13 phosphorylation via ULK1 kinase activity and blocked the autophagic flux induced by amino acid starvation in cells. GW837331X and GW406108X were also reported to inhibit ULK2 with similar activities against ULK1 [7][41].
U-2 exhibited a sub-micromolar IC50 value against ULK1 [11][45]. U-2 decreased the expression level of LC3-II after co-treatment with the lysosomotropic agent chloroquine in HeLa cells under starvation conditions, indicating the blockade of autophagic flux in these cells. U-2 also showed selective cytotoxicity in human liver cancer cells compared to that in normal liver cells. In silico ADMET predicted that the compound U-2 possesses good drug-like properties; however, no experimental evidence has yet been provided for in vitro or in vivo ADME.
Table 1.
ULK1/2 inhibitors as potential anticancer agents.
| Compound | IC50 (ULK1) | IC50 (ULK2) | Cancer (Cell Line) | Synergistic Co-Treatment | |
|---|---|---|---|---|---|
| 1 | SBI-0206965 | 108 nM [1][38] 306 nM [5][22] 130 nM [2][39] |
711 nM [1][38] 3.88 μM [5][22] |
Non-small cell lung cancer (A549, H460) [1][22][38,54] Neuroblastoma (SK-N-AS, SH-SY5Y, SK-N-DZ) [12][46] Renal cell carcinoma (A498, ACHN) [13][47] Leukemia (MV4-11, MOLM-13, HL-60, U937) [14][23][25,55] Breast cancer (MDA-MB-231, MCF-7) [24][56] |
mTOR inhibitor AZD8055 [1][38], daunorubicin [23][55], doxorubicin [24][56], cisplatin [22][54], TRAIL a [12][46] |
| 2 | SBP-7455 | 13 nM [2][39] | Triple negative breast cancer (MDA-MB-468, BT549, MDA-MB-231) [2][39] | PARP inhibitors olaparib, niraparib [2][39] | |
| 3 | Compound 6 | 8 nM [3][21] | - | ||
| 4 | Compound 3 | 120 nM [4][40] | 360 nM [4][40] | - | |
| 5 | MRT67307 | 45 nM [15][24] 170 nM [4][40] 38.2 nM [5][22] |
38 nM [15][24] 230 nM [4][40] 92.3 nM [5][22] |
Leukemia (THP-1, U939, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [25][57] | |
| 6 | MRT68921 | 2.9 nM [15][24] 17.0 nM [5][22] |
1.1 nM [15][24] 20.8 nM [5][22] |
Mesothelioma (M28, REN) [26][58] b Leukemia (REH, MV4-11, MOLM-13, HL-60, U937, THP-1, Molt4, HEL92.1.7, K562, Raji, Jurkat, HL60) [14][17][25][25,49,57] Ovarian cancer (OVCAR3/4/8, COV318/362, CaOV3) [18][50] Various tumors [16][48] |
Carboplatin and pemetrexed [26][58] |
| 7 | Compound 3s | 99.15% inhibition at 10 μM [8][42] c | Lung cancer (A549) [8][42] Lymphoma (U937) [8][42] Breast cancer (HL60) [8][42] Acute myeloid leukemia (MDA-MB-469) [8][42] |
||
| 8 | PF-03814735 | KD 18.1 nM [5][22] d | KD 58.0 nM [5][22] d | Solid tumors [19][20][51,52] | |
| 9 | Hesperidin | KD 16.8 nM [5][22] d | KD 47.3 nM [5][22] d | Various tumors [21][53] | |
| 10 | SR-20295 | 45 nM [9][43] | - | ||
| 11 | ULK-101 | 8.3 nM [6][18] | 30 nM [6][18] | Osteosarcoma (U2OS) [6][18] Non-small cell lung cancer (H838, H727, H2030, A549) [6][18] |
|
| 12 | XST-14 | 13.6 nM [10][44] | 70.9 nM [10][44] | Hepatocellular carcinoma (HepG2, Hep3B) [10][44] | sorafenib [10][44] |
| 13 | GW837331X | 646 nM [7][41] | Similar to ULK1 e [7][41] | - | |
| 14 | GW406108X | 427 nM [7][41] | Similar to ULK1 e [7][41] | - | |
| 15 | U-2 | 0.5 μM [11][45] | Hepatocellular carcinoma (SMMC-7721, HepG2, L02) [11][45] |
a TNF-related apoptosis inducing ligand; b Multicellular spheroids; c IC50 was not determined; d Binding affinity from isothermal calorimetry experiments; e Similar inhibition of ULK1 (IC50 or quantitative inhibitory activity was not reported).