Blood pressure is determined by cardiac output and peripheral vascular resistance. The L-type voltage-gated Ca2+ (Cav1.2) channel in small arteries and arterioles plays an essential role in regulating Ca2+ influx, vascular resistance, and blood pressure. Hypertension and preeclampsia are characterized by high blood pressure. In addition, diabetes has a high prevalence of hypertension. The etiology of these disorders remains elusive, involving the complex interplay of environmental and genetic factors. Common to these disorders are oxidative stress and vascular dysfunction. Reactive oxygen species (ROS) derived from NADPH oxidases (NOXs) and mitochondria are primary sources of vascular oxidative stress, whereas dysfunction of the Cav1.2 channel confers increased vascular resistance in hypertension.
- hypertension
- gestational diabetes
- Cav1.2
1. Introduction
2. Cav1.2 in Vascular Smooth Muscle
2.1. Overview of Ca
v
1.2
L-type Ca2+ (Cav) channels are heteromultimeric complexes comprising pore-forming α1c and auxiliary β, α2δ, and γ subunits [18][27]. The α1c subunit possesses four repeat domains (I–IV) linked by intracellular loops and intracellular NH2-/COOH-termini with each domain containing six transmembrane segments (S1–S6) (Figure 1). The ion-conducting pore is formed by S5 and S6 and the loop between them, whereas the voltage sensor is located in the S4 segments [19][28]. The intracellular COOH terminus, along with intracellular loops, plays important roles in Ca2+-dependent inactivation, channel trafficking, phosphorylation and oxidation [19][20][21][28,29,30]. Cav is activated by membrane depolarization and its activation allows Ca2+ influx through the channel pore. The expression of the α1c subunit itself can form functional channels to conduct Ca2+ ions, whereas the incorporation of auxiliary subunits promotes membrane α1c expression and alters biophysical properties of the channel [19][22][28,31]. Cav channels are sensitive to blockades by dihydropyridines (i.e., nifedipine), phenylalkylamines (i.e., verapamil) and benzothiazepines (i.e., diltiazem) [23][32]. There are four types of Cav channels (Cav1.1–1.4). Cav1.2 is predominantly expressed in the heart and in vascular smooth muscle [24][25][33,34]. Ca2+ influx through the channel is a primary trigger of vasoconstriction and also participates in transcriptional regulation. The auxiliary subunits β2, β3, and α2δ1 are expressed in vascular smooth muscle [26][27][28][29][35,36,37,38]. β subunits, lacking membrane-spanning segments, are located intracellularly and interact with the α interaction domain (AID) on the I-II linker of the α1c subunit [30][39]. The α2δ1 subunit is a single gene product bound together by disulfide bonds. Whereas the α2 subunit is extracellular, the δ subunit contains a single transmembrane segment [31][40]. The diversity of Cav1.2 is also conferred by alternative splicing. Smooth muscle contains Cav1.2 exons 1, 8, 9 *, 31/32, and 33 [32][33][41,42].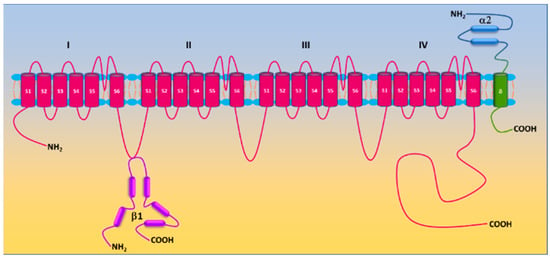
2.2. Regulation of Ca
v
1.2
2.2.1. Regulation by Auxiliary Subunits
The molecular composition of Cav1.2 in vascular smooth muscle cells includes the pore-forming α1c subunit and auxiliary β2/3 and α2/δ1 subunits. The β3 subunit appears to be the principal β isoform in vascular smooth muscle cells [26][35]. Genetic deletion of the β3 subunit resulted in reduced α1c expression in mouse aorta and is associated with a reduction in Ca2+ channel current and a slower inactivation rate [26][35]. This genetic manipulation also attenuates angiotensin II-induced upregulation of Cav1.2 channels in mouse mesenteric arteries and the development of hypertension [29][38].2.2.2. Regulation by Protein Kinases
Protein phosphorylation, a covalent addition of the phosphate group to the side chain of serine, threonine, and tyrosine residues by protein kinases, is a common posttranslational modification to fine tune activities of receptors, ion channels and enzymes. Unsurprisingly, Cav1.2 is a target of protein kinases, and its activity is subject to the regulation by protein phosphorylation. Both α1c and β2 subunits are phosphorylated by protein kinases A (PKA), C (PKC), and G (PKG) [20][34][35][36][37][29,46,47,48,49]. A variety of putative serine/threonine phosphorylation sites have been identified, yet their role in regulating Cav1.2 remains unsettled [38][50]. In vascular smooth muscle cells, the regulation of Cav1.2 by PKA is controversial. PKA is found to either inhibit or enhance Cav1.2 activity [39][40][41][42][43][44][45][51,52,53,54,55,56,57]. The stimulatory effect of PKA on Cav1.2 in vascular smooth muscle cells depends on anchoring adenyl cyclase 5 and PKA by A-kinase anchoring protein 150 (AKAP150) to the proximity of Cav1.2 [44][46][56,58]. Phosphorylation of Ser 1928 in the COOH-terminus of the Cav1.2 α1c subunit is required for PKA-stimulated channel activity in vascular smooth muscle cells [44][56]. Activation of PKG exhibits inhibitory effects on vascular Cav1.2 [40][45][47][48][52,57,59,60]. PKG mediates nitric oxide-induced inhibition of Cav1.2 [49][50][61,62]. Activation of PKC by phorbol esters and by Gq-coupled receptors also potentiates vascular Cav1.2 activity [51][52][53][54][55][56][57][58][63,64,65,66,67,68,69,70]. Basal Cav1.2 activity is evidently under the tonic control of PKC as PKC inhibition/PKCα depletion enhances Cav1.2 activity in vascular smooth muscle cells [55][67]. Activation of PKG by nitric oxide (NO) suppresses Cav1.2 activity [37][49]. Similar to PKA, PKC is anchored by AKAP150 to adjacent Cav1.2 to alter channel activity [59][71]. Protein tyrosine kinase c-Src promotes tyrosine phosphorylation of the α1c subunit and enhances Cav1.2 activity, which is believed to participate in regulating smooth muscle contractility [60][72]. c-Src via its SH2 and SH3 domains binds to the COOH-terminus of the α1c subunit [58][70]. Y2122 in the COOH-terminus of the α1c subunit appears to be the major phosphorylation site of c-Src [58][61][70,73]. In vascular smooth muscle cells, c-Src enhances Cav1.2 activity [52][62][63][64][64,74,75,76]. Phosphoinositide 3-kinases (PI3Ks) are found to increase Cav1.2 activity in vascular smooth muscle cells [65][66][77,78]. PI3Kγ potentiates Cav1.2 activity by facilitating plasma membrane translocation of α1c subunits and this effect is mediated by AKT/PKB-induced β2 subunit phosphorylation [67][68][69][70][79,80,81,82]. Integrins also participate in the mechanotransduction process of pressure-induced myogenic tone [71][83]. Integrin receptor activation is found to increase Cav1.2 activity via c-Src and PKA [72][73][74][84,85,86].2.2.3. Regulation by Small GTPases
The Ras superfamily of small GTPases (also known as small G-proteins) are cellular switches that regulate a variety of biological processes in living cells. They have been implicated in regulating Ca2+ homeostasis and Cav1.2 is recognized as an important effector of the RGK subfamily (Rem, Rem2, Rad, and Gem/Kir) [75][76][87,88]. Unlike vascular Cav1.2, direct phosphorylation of the cardiac Cav1.2 α1c subunit does not contribute to PKA-stimulated channel activity as mutating all PKA consensus sites in the α1c subunit fails to block the increase in Cav1.2 activity in response to β-adrenergic stimulation [77][78][89,90]. Cardiac Cav1.2 is under tonic inhibition of Rad due to its association to both α1c and/or β subunits [79][80][91,92]. β-adrenergic stimulation induces RAD phosphorylation, promotes the release of RAD from Cav1.2, and increase channel activity [78][90].2.3. Ca
v
1.2 and Myogenic Tone
Vascular smooth muscle cells in resistant arteries and arterioles possess intrinsic ability to contract in response to an increase in intraluminal pressure and to relax upon a decrease in intraluminal pressure [81][97]. This phenomenon is defined as myogenic response and the steady-state of vascular smooth muscle cell contractility in these vessels is termed as myogenic tone. Myogenic tone sets the basal vascular tone and distribution of blood flow to and within tissues/organs. In principle, peripheral vascular resistance can be described by the Poiseuille equation: R = 8Lη/πr4, where R is the resistance, L is length of the vessel, η is viscosity of blood, and r is the radius of the vessel. According to the Poiseuille’s law, peripheral vascular resistance is inversely proportional to the radius to the fourth power. Vascular smooth muscle cells in resistance arteries/arterioles become depolarized in response to increased intraluminal pressure [82][83][84][98,99,100]. Various mechanisms have been proposed to regulate myogenic tone. It is widely believed that pressure-induced membrane depolarization in vascular smooth muscle cells is instigated by mechanosensitive or stretch-activated cation channels including transient receptor potential (TRP) channels and epithelial Na+ channels (ENaCs) [85][86][87][88][89][90][91][101,102,103,104,105,106,107]. The membrane depolarization leads to increased [Ca2+]i and vasoconstriction [83][92][99,108]. The increases in both [Ca2+]i and/or myogenic tone are blocked by the removal of extracellular Ca2+ or by Cav1.2 blockers [83][93][94][95][96][97][98][99][99,109,110,111,112,113,114,115]. These findings suggest that altered intraluminal pressure initiates myogenic tone via membrane depolarization and subsequent opening of Cav1.2. This notion is corroborated by findings from the genetic deletion of Cav1.2 α1c subunit in smooth muscle [100][116]. Such a manipulation abolishes myogenic reactivity in murine tibialis arteries [100][116].3. Roles of Cav1.2 in the Pathogenesis of Hypertension-Related Disorders
3.1. Aberrant Vascular Tone in Hypertension-Related Disorders
Hypertension is associated with increased peripheral vascular resistance. As myogenic tone is the fundamental element of vascular tone, it is reasonable to speculate that myogenic tone is altered in hypertension-related disorders. In patients with hypertension, myogenic tone is increased in coronary arterioles [94][110]. Increased myogenic tone is also noted in resistance arteries from the adipose tissue of paravertebral muscles of hypertensive patients [101][130]. Commonly used rat models of experimental hypertension include the spontaneously hypertensive rat (SHR), stroke-prone spontaneously hypertensive rat (SHRSP), Milan hypertensive strain (MHS), vasopressin-deficient (Di/H) rats, two-kidney, one-clip (2K1C rats), and among others. Compared to the Wistar-Kyoto rat (WKY), myogenic tone of afferent arterioles and arcuate arteries from SHR kidneys is increased [102][103][131,132]. Mesenteric arteries of SHR and MSH also exhibits higher myogenic tone than those of WKY and Milan normotensive strain (MNS), respectively [104][105][133,134]. Elevated myogenic tone is observed in cerebral arteries/basilar arteries of SHR/SHRSP/Di/H compared to WKY and Di normotensive (Di/N) rats, respectively [106][107][108][109][135,136,137,138]. Myogenic tone is increased in skeletal muscle resistance arteries of diabetic patients [110][144]. Various animal models such as type I diabetic rodents induced by streptozotocin (STZ), obese/type II diabetic rodents induced by high-fat diet (HFD), type II diabetic HFD/STZ rodents, type II diabetic Goto-Kakizaki (GK) rats, and type II diabetic C57BL/KsJ-db/db mouse have been developed in diabetes research [111][112][145,146]. Systemic vascular resistance (also known as total vascular resistance) and arterial blood pressure are increased in C57BL/KsJ-db/db mice, HFD rats, and GK rats [113][114][115][116][117][118][119][120][121][147,148,149,150,151,152,153,154,155]. Mesenteric arteries of HFD mice, HFD/STZ mice, and diabetic (db/db) mice displayed higher myogenic tone compared to their counterparts [46][110][121][122][58,144,155,156].3.2. Dysfunction of Vascular Ca
v
1.2 in Hypertension-Related Disorders
As aforementioned, activation of Cav1.2 in vascular smooth muscle cells is essential for the development of myogenic tone [83][84][93][94][97][98][100][99,100,109,110,113,114,116]. The enhanced myogenic tone in peripheral resistance arteries and arterioles in hypertension-related disorders suggests potential dysfunction of vascular Cav1.2. Indeed, this notion is substantiated by lines of evidence from functional studies. First, the increased myogenic tone in resistance arteries/arterioles of hypertensive and diabetic animals was normalized by the Cav1.2 blocker nifedipine [46][103][123][58,132,181]. Whereas specific deletion of the mineralocorticoid receptor reduces KCl- and Cav1.2 agonist Bay K 8644-induced vasoconstriction of mesenteric arteries [124][125][142,182], Bay K 8644-induced contraction and increase in [Ca2+]i in 2K1C rat aorta is greater than in the control 2K rats [126][183]. Animal models of hypertension-related disorders have provided mechanistic insights into the understanding of the Cav1.2 dysfunction in these disorders. Both aberrant expression of and dysregulation of Cav1.2 contribute to vascular Cav1.2 dysfunction. Various studies reveal increased protein expression of α1c [28][29][127][128][129][37,38,187,192,193], α2δ1 [28][128][37,192], and β3 [28][29][37,38] subunits in mesenteric, femoral, and cerebral arteries of SHR and angiotensin II-infused C57BL/6 mice. Consistently, increased expression of Cav1.2 is associated with enhanced channel activity in vascular smooth muscle cells [55][129][130][131][132][133][134][67,185,193,194,195,196,197]. The expression and activity of Cav1.2 are reduced in mesenteric arteries of aged mice lacking mineralocorticoid receptors in smooth muscle cells [135][143]. Dexamethasone administration increases the expression of the cardiac Cav1.2 α1c subunit in rats [136][198]. As expected, Cav1.2 activity in A7r5 cells is increased following chronic dexamethasone exposure [137][199]. An increase in protein expression of the α1c subunits is also observed in cerebral arteries of STZ rats, which is associated with increased Cav1.2 activity and contraction to KCl [138][203]. Cav1.2 also displays increased activity in vascular smooth muscle cells of cerebral and mesenteric arteries from STZ, GK, and HFD rats and db/db mice [119][139][140][141][153,204,205,206]. Human arteries from diabetic patients have higher Cav1.2 activity due to increased phosphorylation of α1C at Ser1928 by PKA [44][56]. Gal-1 is highly expressed in the placenta [142][207]. Circulating Gal-1 level also increases during gestation [143][208]. Given the critical role of Gal-1 in regulating Cav1.2 surface expression/activity discussed above, it is reasonable to speculate that the elevated Gal-1 in pregnancy may contribute to reduced uterine arterial myogenic tone by suppressing Cav1.2 surface expression/activity [144][145][209,210].4. Roles of Reactive Oxygen Species (ROS) in the Pathogenesis of Hypertensive Disorders
4.1. Overview of ROS
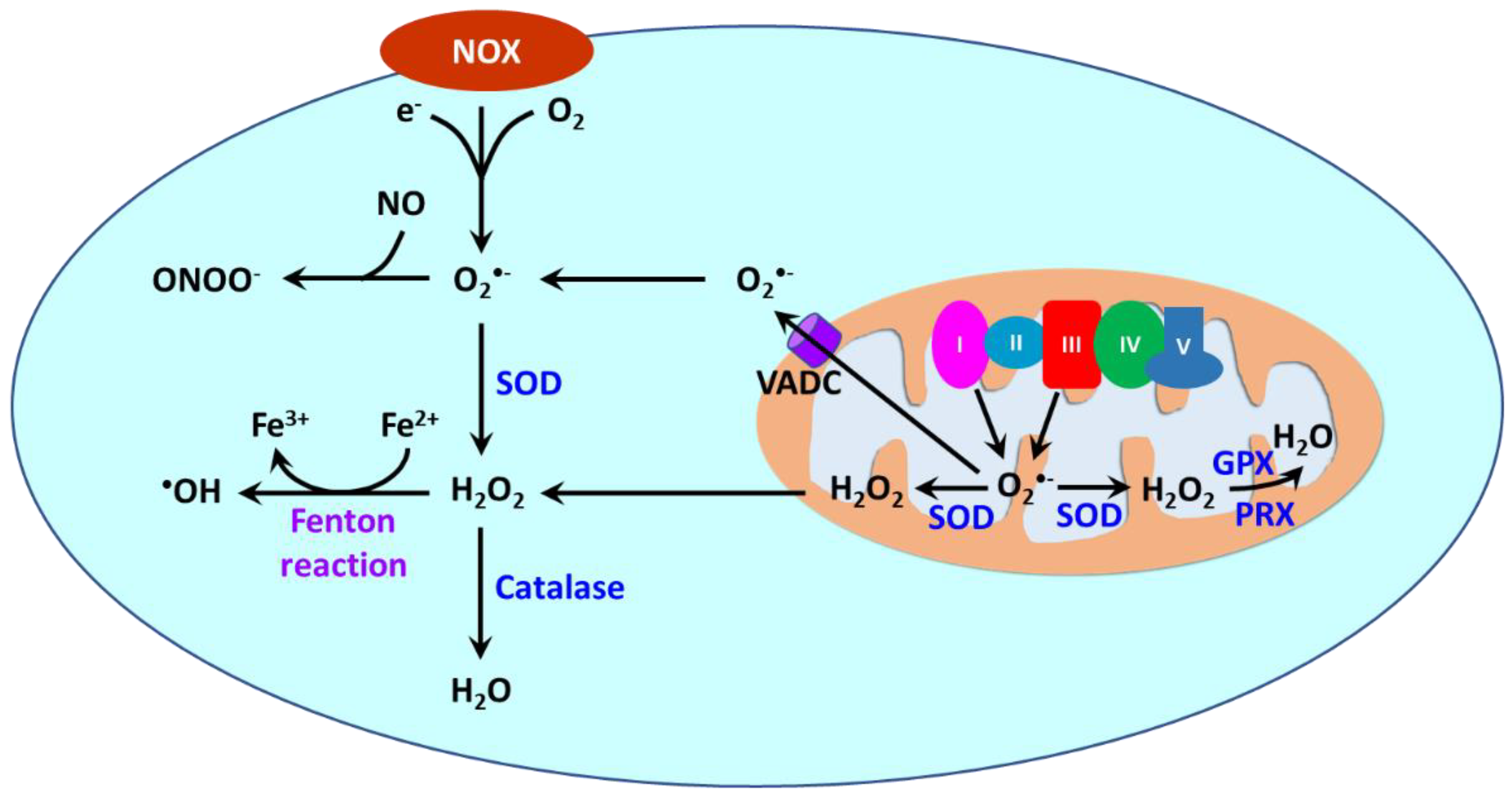
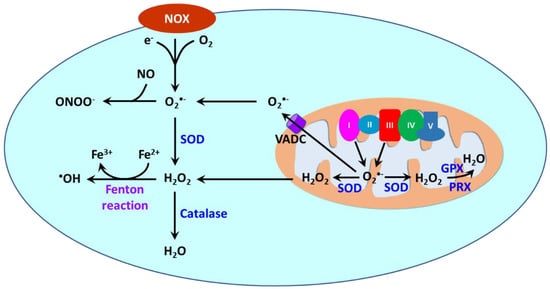
Reactive oxygen species (ROS) are oxygen-containing molecules naturally produced in cellular metabolism. ROS comprise free radicals such as superoxide anion (O2•−) and hydroxyl radical (•OH) and nonradical molecule hydrogen peroxide (H2O2). O2•− is formed from the one-electron reduction of molecular oxygen (O2). It is a precursor to a cascade of other ROS. Its dismutation, either occurring spontaneously or being catalyzed by superoxide dismutases (SODs), produces H2O2. Through the Fenton reaction, H2O2 can be reduced to •OH. Moreover, the reaction between O2•− and nitric oxide (NO) results in the formation of peroxynitrite (ONOO−). ROS are produced in different cellular compartments including mitochondria, endoplasmic reticulum (ER), lysosomes, peroxisomes, and plasma membrane [14][146][147][23,213,214]. Nicotinamide adenine dinucleotide phosphate oxidases (NOXs) and mitochondria are the major sources of ROS [148][215] (Figure 23). NOXs are a family of transmembrane proteins that catalyze the transfer of electron donated by NADPH to molecular oxygen to form O2•− [149][216]. NOXs comprise seven isoforms (NOX1-5 and dual oxidases (DUOX) 1 and 2). NOX1, NOX2, NOX4, and NOX5 are primary isoforms in vasculature [150][217].