Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Aniello Meoli and Version 2 by Rita Xu.
Cystic fibrosis (CF), the most common genetically inherited disease in Caucasian populations, is a multi-systemic life-threatening autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (
CFTR
) gene. In 2012, the arrival of
CFTR
modulators (potentiators, correctors, amplifiers, stabilizers, and read-through agents) revolutionized the therapeutic approach to CF.
- CFTR
- CFTR modulators
- cystic fibrosis
- inflammation
1. Introduction
Cystic fibrosis (CF), the most common genetically inherited disease in Caucasian populations, is a multi-systemic life-threatening autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on the long arm of chromosome 7 [1]. This gene encodes for the CFTR protein, a cyclic adenosine monophosphate (cAMP)-regulated channel classically responsible for chloride (Cl−) and bicarbonate (HCO3−) passive transport across epithelial surfaces, crucial in maintaining epithelial surface hydration and regulating luminal pH, in turn fundamental for epithelial barrier function and innate defense [1][2][1,2]. A defective CFTR function strongly affects epithelial homeostasis, especially in the respiratory system, which determines much of the morbidity and mortality in CF patients, and gastrointestinal tracts.
In the lungs of people with CF (PWCF), the impaired ion transport derived from the CFTR dysfunction results in the dehydration and hypersecretion of mucus that contributes to airway obstruction and chronic bacterial colonization with recurrent pulmonary exacerbations, especially by Pseudomonas aeruginosa; furthermore, the exaggerated inflammatory response, hallmark of this disease, in synergy with an impaired resolution of inflammation, leads to progressive lung damage with bronchiectasis formation, wall thickening, and lung function decline. Although recurrent respiratory exacerbations represent a considerable stimulus for the promotion of the chronic inflammatory state that characterizes CF, increasing evidence suggests that CFTR dysfunction itself drives a dysregulated inflammatory response and that, before any infection, CF airways are already a perfect milieu for the amplification of the immune-inflammatory cascade [3]. The aforementioned alterations are present since early childhood and are accompanied by increasing colonization, first dominated by Haemophilus influenzae and Staphylococcus aureus and then by P. aeruginosa, and to a lesser extent Burkholderia cepacia complex (BCC) and Stenotrophomonas maltophilia [4][5][4,5].
Regarding the gastrointestinal system, meconium ileus at birth, distal intestinal obstruction syndrome (DIOS), and constipation are an interrelated group of obstruction syndromes with variable severity that characterize PWCF [6].
In CF, pulmonary and gastrointestinal complications are also associated with pancreatic insufficiency, diabetes, malabsorption with malnutrition, liver disease, and infertility [7]. Furthermore, an altered microbiome plays a role in disease progression, as demonstrated by the consequences of profound dysbiosis, both pulmonary and intestinal, that characterize this disease [8][9][10][8,9,10].
The recent evidence of the expression of CFTR in immune cells, combined with the well-known remarkable neutrophilic infiltration of the lungs of PWCF, sparked new studies on the role of phagocytes in the chronic inflammatory and impaired immune response that characterize CF, shifting the perspective from a rather epithelium-centric vision of this disease to a wider scenario [3][11][12][13][14][15][3,11,12,13,14,15].
In 2012, the arrival of CFTR modulators (potentiators, correctors, amplifiers, stabilizers, and read-through agents) has revolutionized the therapeutic approach to CF especially with the introduction of triple combination therapy (elexacaftor/tezacaftor/ivacaftor [ELX/TEZ/IVA], Trikafta and Kaftrio in United States and Europe, respectively) in 2019. This therapy, known also as highly effective modulator therapy (HEMT), realized a real paradigm shift in the management of CF as these molecules target the upstream underlying defect of the disease permitting the treatment of patients with almost one copy of F508del-CFTR, the most common CF-causing mutation (found in ~90% of PWCF) [2][11][2,11].
2. The Role of CFTR Dysfunction in Phagocytes: A Paradigm Shift
2.1. Monocyte/Macrophages
Macrophages represent the first line of defense against pathogens in the lungs and are divided into tissue-resident and recruited (monocyte-derived macrophages [MDMs]); tissue-resident macrophages are further divided in alveolar (which reside in the airway lumen) and interstitial (located in the lung parenchyma) [16][31]. Alveolar macrophages are the dominant immune cell in the steady state and conduct several functions: ensure and modulate the inflammatory response, either directly or by the crosstalk with the adaptive immune system, actualize the bacterial killing through a complex mechanism called phagocytosis, maintain tissue homeostasis acting as scavenger cells, and participate in regeneration processes [17][18][32,33]. The ability to promote all these functions is due to their plasticity. In 2000, Mills et al. distinguished macrophages in M1/kill and M2/repair profile on the basis of the pathway they utilize to metabolize arginine; specifically, M1 metabolize arginine to nitric oxide, an inhibitor of proliferation, whereas M2 produce ornithine, a promoter of proliferation. Macrophages are able to switch between classes, and such plasticity is important in the macrophage’s ability to regulate the acute inflammatory response [19][20][34,35]. Whether macrophages display an M1 or M2 phenotype is dependent upon the tissue milieu. M1 responses are associated with IL-6, IL-8, IL-12, and TNF-α production and cell surface expression of CD80 or CD86, which attract neutrophils and stimulate Th1 responses and further M1-type activation. On the contrary, M2 responses are linked with TGF-β, VEGF, EGF, IL-4, and IL-13 production, cell surface expression of CD163 or CD206, and the propensity to stimulate Th2 responses such as antibody production and further amplification of M2-type activation. Appropriate activation of the inflammatory response and subsequent resolution requires a balance between macrophage subpopulations [18][20][21][33,35,36]. Despite the clear predominance of neutrophilic infiltration, in the airways of PWCF, the absolute number of macrophages is not only increased during lung exacerbations but even from the later stages of fetal development and in young children with CF without detectable infection [22][23][24][37,38,39]. Furthermore, as demonstrated by Meyer et al., CF macrophages display both an augmented M1 profile, responsible for a proinflammatory phenotype with high levels of IL-8, IL-6, and TNF-α and low levels of IL-10, and an exaggerated M2 profile that might contribute to remodeling processes and fibrotic changing typical of CF. In addition, the authors concluded that Azithromycin regulates profile shifting by inhibiting M1 polarization and favoring the M2 profile [25][40]. However, bronchoalveolar lavage (BAL) obtained from CF lungs contains large concentrations of TNF-α, IL-1β, IL-6, and IL-8, but little IL-10, a pattern associated with M1 polarization [20][35]. Tarique et al. clarified these findings, obtaining MDMs from people with CF and healthy volunteers, and demonstrating that CF M2 polarization was dysfunctional as evidenced by the decreased ability to perform endocytosis [26][41]. On the other hand, CF M1 macrophages showed a decreased phagocytic activity and the inability to switch to the M2 phenotype with a consequent imbalance of the immune-inflammatory cascade towards a pro-inflammatory state [26][41]. Yoshimura et al. in 1991 demonstrated that CFTR is expressed not only in epithelial cells that strongly influence the lung milieu but also in human macrophages and neutrophils [27][42]; moreover, in macrophages, it has been detected both at the surface and intracellularly [28][43]. Mutations in the CFTR gene can affect macrophage function with different effects based on mutation type and in different phases of phagocytosis. In CF, the impaired macrophage function is not due exclusively to the altered epithelial environment, a hallmark of the disease, but also to a direct effect on macrophages [28][43]. The involvement of macrophages in the creation of the proinflammatory status typical of CF was demonstrated by Bruscia et al. in their work [29][44]. They compared the cellularity and cytokine levels in the BAL of wild-type (WT), heterozygous (HET), and CF mice after P. aeruginosa LPS exposure using a murine in vivo model of inflammation. The lungs of CF mice showed a more robust neutrophil infiltration with higher levels of macrophage-derived cytokine that promote their migration (KC, MIP-2, IL-8) and survival (G-CSF) with a slower neutrophil clearance in comparison with WT lungs. Furthermore, the BAL of CF mice was characterized by higher concentrations of macrophage-derived cytokine that stimulate the acute inflammatory response (IL-1α, IL-6) and innate immunity (IL-12) and lower levels of IL-10 (a cytokine with anti-inflammatory function) in comparison with WT mice. In addition, authors found that HET mice and cells have a phenotype between WT and CF, and thus, a single allelic CFTR mutation is sufficient to augment proinflammatory activation in response to LPS in CF, implying CFTR-dependent defects in CF macrophages [29][44]. Moreover, the direct role of CF macrophages in exacerbating the immune-inflammatory cascade was demonstrated by transplanting the bone marrow of WT into CF mice. Indeed, authors highlighted that CFTR+/+ myeloid cells, including macrophages, significantly reduce the inflammatory response to P. aeruginosa LPS in CFTR−/− animals [16][29][31,44]. Bonfield et al. reached the same conclusions using an in vivo mouse model, showing that defective myeloid CFTR contributes to increased inflammation with elevated cytokine production, the recruitment of neutrophils, and the inability to resolve infection even in presence of functional epithelial CFTR [30][45]. However, in the case of CFTR dysfunction, the macrophage function is not only affected at the level of the promoted altered inflammatory response but also at the level of the phagocytosis and autophagy processes [17][31][32,46]. When a pathogen is encountered, alveolar macrophages initiate the immune response releasing cytokines and chemokines and recruiting neutrophils and monocyte-derived macrophages (MDMs) that drive inflammation; specifically, MDMs are recruited through the release of monocyte chemoattractant protein-1 (MCP-1/CCL2) by macrophages that can also impact leukocyte behavior, influencing adhesion, polarization, effector molecule secretion, autophagy, killing, and survival [17][32][32,47]. In response to pathogens, macrophages also initiate the phagocytosis process that involves several phases: (i) detection of the particle to be ingested, (ii) activation of the internalization process, (iii) formation of a specialized vacuole called a phagosome, and (iv) maturation of the phagosome to transform it into a phagolysosome [33][48]. Macrophages sense pathogens by recognizing pathogen-associated molecules (PAMPs), such as lipopolysaccharide (LPS), flagellin, and dsRNA, through receptors known as pattern-recognition receptors (PRRs), expressed also on immune and structural cells, including airway epithelium. Furthermore, PRRs recognize the damage-associated molecular patterns (DAMPs), cytoplasmic and nuclear components such as HMGBI, ATP, and adenosine that are released into the extracellular environment by damaged cells [16][31]. There are several types of PRRs, but TLRs have been studied most intensely in CF. In non-CF patients, when exposed to P. aeruginosa, macrophages through the TLR4 and TLR5, expressed on their surface, initiate the pro-inflammatory cascade that activates NF-κB and MAPK signaling. On the CF macrophage surface, TLR4 is increased, whereas TLR5 is decreased, leading to excessive pro-inflammatory signaling [20][35]. Studies suggest CF macrophages have defective bacterial killing, but the underlying mechanisms are not fully understood [20][35]. Di et al. in their study were the first to associate the defective CFTR function with failed alveolar macrophage lysosomal acidification, a crucial process for efficient bactericidal activity. Indeed, they showed that CFTR protein participates in phagosomal pH control and therefore its dysfunction leads to a defective killing of internalized bacteria although the retained ability to phagocytose and generate an oxidative burst [34][49]. Deriy et al. reached a similar conclusion finding a tight correlation between CFTR genotype and levels of lysosomal acidification in alveolar macrophages [35][50]. Nevertheless, these data were subsequently confuted by two studies that showed that phagolysosomal acidification in alveolar macrophages is not dependent on CFTR channel activity [36][37][51,52]. More recently, similar results were highlighted by Law et al. on MDMs using optical nanosensors, a novel means that accurately measures macrophage phagolysosomal pH, although this finding has to be confirmed on alveolar macrophages using the same technique [38][53]. However, a study conducted by Riazanski et al. demonstrated that drugs are able to facilitate the activity of transient receptor potential canonical-6 (TRPC6) channel potentiating phagosome acidification and the bacterial-killing of CF macrophages [39][54]. The major difficulty in studying the impact of a dysfunctional CFTR on the bacterial-killing function of CF macrophages lies in the reproducibility of the observations, strongly influenced by the sensitivity of these cells to minimal changes in the environment of culture or purification. In addition, the complex cell signaling promoted in macrophages by live bacteria is extremely difficult to reproduce with opsonized beads [16][31]. Furthermore, CFTR−/− cell lines used in some studies as a surrogate for F508del cells represent a possible bias due to the lack of unfolded protein response (UPR); indeed, F508del mutation causes misfolding of the CFTR protein triggering the UPR, involving endoplasmic reticulum (ER) stress, and NF-κB activation [40][55]. Some studies have focused on the killing of specific pathogens by CF macrophages. A recent study demonstrated that they have impaired lysosomal degradative capacity of B. cenocepacia, a highly virulent member of BCC, which resides in LC3-labeled autophagosomes but not of Escherichia coli, which are enclosed in vacuoles that do not acquire LC3 [41][56]. Regarding the impaired killing of P. aeruginosa by CF macrophages, both defective phagocytosis function and activation of autophagy might contribute to this phenomenon. Indeed, Caveolin-1, which mediates P. aeruginosa internalization, is expressed at low levels in activated CF macrophages [16][42][43][31,57,58]. Moreover, autophagy, a fundamental mechanism of cytoplasmic protein turnover, has been directly linked to the ineffective bactericidal function in CF macrophages. Specifically, it is defective in CF airways because of the depletion of the autophagy-related protein Beclin 1 (BECN1) with consequent accumulation of SQSTM1/p62 substrate which promotes a pro-inflammatory status sequestering misfolded ubiquitinated F508del-CFTR and anti-inflammatory proteins, such as PPARγ and IK-Bα. In their study, Ferrari et al. demonstrated that the proteostasis regulator cysteamine, which restores the function of F508del-CFTR mutant, reestablished the autophagy mechanism of CF MDMs, restoring both bacterial internalization and clearance of P. aeruginosa through a process that involves upregulation of BECN1 [16][17][31][31,32,46]. In addition to acidification defects, CF macrophage phagosomes also have alterations in NADPH oxidase assembly and subsequent ROS production based on a decreased activation of cytosolic NADPH oxidase components, such as p47phox and p40phox, crucial for complex formation. This evidence, independent of pathogens but amplified by B. cenocepacia, results in an impaired bacterial killing with intracellular growth of bacteria such as B. cenocepacia, P. aeruginosa, and Mycobacterium abscessus [16][44][45][46][31,59,60,61]. Another alteration that might influence innate response was reported by Wright et al. who reported a failed expression of scavenger receptors such as the mannose receptor (CD206) and the macrophage receptor with collagenous structure (MARCO) by CF macrophages with consequent impaired binding and internalization of unopsonized particles as well as microbes resulting in dysfunctional phagocytosis [47][48][62,63]. Finally, given the key role of macrophages in extracellular iron depletion, an impairing in their iron-related protein expression profile in CF, as showed by Hazlett et al., might contribute to the elevated iron levels found in CF sputum and BAL fluid. Specifically, in their study authors reported reduced ferroportin (Fpn) and augmented transferrin receptor 1 (TfR1) levels in CF MDMs compared to non-CF MDMs with consequent advantage for pathogens such as P. aeruginosa, in whose metabolism iron plays a crucial role [49][64].2.2. Neutrophils
Polymorphonuclear neutrophils (PMNs) play a central role in host defense against microbes and are early mobilized during an inflammatory response, both infectious and noninfectious, from the bone marrow, where they mainly reside [50][65]. In case of infectious stimulus, once arrived at the inflammation site driven by AECs and macrophage-derived cytokine and chemokine, they produce ROS, secrete antimicrobial peptides via the degranulation mechanism, eliminate microorganisms through phagocytosis process, and trap bacteria in neutrophil extracellular traps (NETs) endowed with antimicrobial activities [20][51][35,66]. Neutrophils, besides their involvement in primary host defense, also contribute to the regulation of immune responses, including the amplification of inflammatory cascade. Indeed, they produce pro-inflammatory cytokines such as IL-1, IL-6, IL-7, IL-17, IFN-γ, and TNF-α [52][67]. During phagocytosis, they confine pathogens in the phagosome and release into it different bioactive agents. These molecules are divided into two categories based on their provenience: proteins synthesized in myeloid precursors during granulopoiesis and stored in granules (serine proteases [azurocidin, proteinase-3, cathepsin G, and elastase], MMP [MMP-9], MPO, defensins, and lactoferrin) and ROS, produced de novo at the time of PMN activation by the NADPH oxidase [50][53][54][65,68,69]. In neutrophils, granules are divided into at least four different types: (i) primary granules, also known as azurophilic granules, that are the main storage site of the most toxic mediators; (ii) secondary granules, also known as specific granules; (iii) tertiary granules, that like secondary ones contain lactoferrin and MMP-9; and (iv) secretory vesicles that contain human serum albumin. These granules are also released during the degranulation process, strictly controlled by different pathways in turn activated by receptors in the plasma membrane or by the phagosomal membrane [55][70]. After the elimination of the pathogen, the cessation of the inflammatory response is achieved through the apoptosis of recruited neutrophils [50][65]. In case of failed eradication of pathogens, neutrophils continue to release their arsenal of destructive enzymes and amplify the immune-inflammatory cascade, especially in diseases such as CF, in which the epithelial lung milieu is impaired in a proinflammatory sense. In CF, neutrophils are the predominant immune cells infiltrating the airway mucosa and the intralumenal space of bronchioles (accounting for ~70% of the total cell count in BAL fluid) driven by IL-8 and IL-17 secretion; their load as well as the extracellular activity of the protease NE in BAL fluid correlates well with disease progression in CF patients, from infancy to adulthood [56][57][58][71,72,73]. In normal homeostatic conditions, neutrophils are short-lived and undergo spontaneous apoptosis to guarantee the termination of the inflammatory insult but, similarly to macrophages, are also characterized by remarkable plasticity. Indeed, although when they leave the bone marrow neutrophils have already a default pro-apoptotic program, it can be modified by stimuli received in the target tissue including IL-8 and GM-CSF, that promote their persistence and longevity [56][59][60][71,74,75]. IL-8, also known as CXCL8, is mainly produced by AECs and macrophages, and one of its major functions is to attract and activate neutrophils and delay their apoptosis via their IL-8 receptor (mainly CXCR2) [61][62][76,77]. The elevated levels of IL-8 that characterize CF, given its anti-apoptotic action on neutrophils, contribute to determining the remarkable neutrophilic infiltration of the CF lung. Roussel et al. demonstrated that a dysfunctional CFTR leads to enhanced IL-8 synthesis upon exposure to P. aeruginosa because of the action of multiple TLRs acting redundantly. Furthermore, the decreased level of extracellular glutathione present in CF with consequent higher sensitivity to ROS further results in an increased production of IL-8 via the function of NADPH oxidase [61][63][76,78]. In CF, in addition to IL-8/IL8R derived signaling, other pathways such as the CXCR4/CXCL12 axis and the oxygen-dependent transcription factor hypoxia-inducible factor-1α (HIF-1α) might concur in prolonging the lifespan of neutrophils consequently increasing the neutrophilic infiltration [61][64][65][76,79,80]. Figure 1 summarizes the involvement of phagocytes and neutrophils in driving lung disease progression in CF.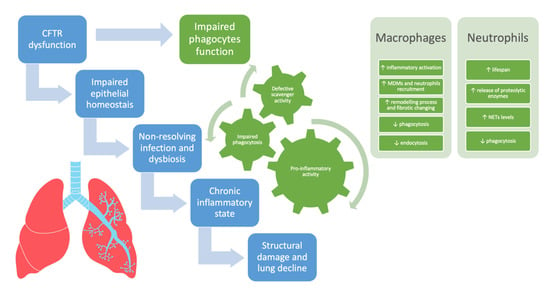
Figure 1. The involvement of phagocytes and neutrophils in driving lung disease progression in cystic fibrosis. CFTR = cystic fibrosis transmembrane conductance regulator, MDMs = monocyte-derived macrophages, NETs = neutrophil extracellular traps.