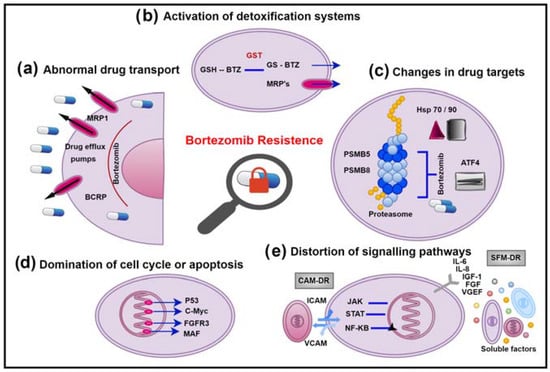
Resistant cells of cancer patients that do not respond to chemotherapy have highly expressed ATP-Binding Cassette (ABC) transporter proteins located in the cytoplasmic region of their membranes
[1][53]. ABC transporters are responsible for transporting drugs and drug metabolites in the organism, working as ATP dependent (
Figure 1a). The ABC protein family has at least 48 known members in humans, most of which are drug transporters
[2][54]. A high expression of ABC transporters has been shown to be responsible for MDR
[3][55]. The most studied efflux transporters are ABCB1 (P-glycoprotein), ABCG2 (BCRP), LRP, and MRP1-9. P-glycoprotein (P-gp), the first discovered member of the ABC transporter family, is encoded by the MDR-1 gene
[4][56]. P-gp is the primary drug transporter protein, which binds the drug and carries it against a concentration gradient by ATP hydrolysis
[5][57]. The expression of P-gp in healthy human tissues functions as a natural detoxification mechanism for excreting drugs and other xenobiotics from the body. P-gp is also overexpressed in cancer cells, resulting in a decrease of the intracellular drug concentration by inhibiting the uptake of many structurally different drugs into cells and extruding them from tumour cells
[6][58]. Since most of the routinely used anticancer agents are substrates of P-gp, cancer cells with higher levels of P-gp can develop resistance during adaptation to the treatment
[7][35]. A total of 6% of newly diagnosed MM patients are P-gp positive, while more than 43% are P-gp positive after chemotherapy
[8][28]. In patients with MM, P-gp expression is usually increased after bortezomib, and some studies indicated that Bortezomib is a poor substrate for P-gp
[9][59]. In addition, other studies reported that Bortezomib could reduce P-gp expression in MM cells
[10][60].
Figure 1. Bortezomib resistance mechanisms in MM. (a) Abnormal drug transport. (b) Activation of detoxification systems. (c) Changes in drug targets. (d) Domination of cell cycle or apoptosis. (e) Distortion of signalling pathways.
Breast Cancer Resistance Protein’s (BCRP) role in normal tissues, similar to P-gp, is preserving the organism as the first line of defence against toxins. It was initially discovered in anthracycline-resistant MCF-7/AdVrp human breast cancer cells
[11][61]. BCRP is prominently expressed in the placenta, small intestine and colon epithelium, liver canalicular membranes, and breast tissue
[12][62]. Increased expression of BCRP was noticed in many drug resistant tumour cell lines
[13][63]. Altered expression in MM cells was associated with drug resistance and poor prognosis
[14][29]. However, BCRP is more expressed in MM stem cells, leading to disease relapse
[15][64].
Lung Resistance-related Protein (LRP) is also called major vault protein (MVP or VAULT1). It was first detected in drug-resistant lung cancer cell lines
[16][65]. Vaults are ribonucleoprotein particles comprising RNA and protein and are found in the cytoplasm as a fraction of the nuclear membrane and nuclear pore complex
[17][66]. Thus, they contribute to drug resistance by transporting substances between the nucleus and cytoplasm. LRP is widely distributed in normal tissues and overexpressed in drug-resistant tumour cells
[18][67]. Overexpression of LRP was reported in leukaemia
[19][68], testicular tumours
[20][69], and breast cancers
[21][70]. In MM, the expression of LRP was observed in patients treated with Melphalan
[22][71] and Bortezomib
[14][29].
The MRP family responsible for MDR includes nine members (MRP-1, MRP-2, MRP-3, MRP-4, MRP-5, MRP-6, MRP-7, MRP-8, MRP-9). MRP-1 is the first member of the family and is expressed in various organ and cell types
[23][72]. The tissue distribution of MRP-1 limits the penetration of certain cytotoxic agents and MRP-1 thus contributes to pharmacological barriers in the body
[24][73]. MRP-1 can carry structurally different kinds of glutathione (GSH) conjugated organic anions
[25][74]. GSH is required for resistance because many studies showed that drug transport occurs only in the presence of reduced GSH
[26][27][75,76]. MRP-1 has various complex interactions with GSH and GSH, and thus appears to be co-transported with (or cross-stimulates transport of) the drug
[28][77]. Until now, some studies have reported that MRP-1 expression level is high in resistant MM cells
[7][29][30,35], while others have reported the opposite
[10][60]. MRP-2 is similar to MRP-1 in its ability to confer resistance to a spectrum of anticancer drugs in vitro
[30][31]. MRP-3 expression appears to play a role in compensating for the loss of MRP-2 in liver diseases
[31][32]. A high expression of the MRP-6 gene in resistant tumour cells was found only in cell lines highly expressing the MRP-1 gene
[32][34]. MRP-7 can also carry a large proportion of organic anions in vitro, and it was reported that MRP-7 contributed to various anticancer agents in drug resistance
[7][33][35,78]. It was suggested that MRP-8 could be a biomarker for predicting the treatment outcomes of AML
[34][36]. Drug resistance caused by ABC transporters is important for MM as in all diseases. Bortezomib is the most potent drug for the treatment of MM, and 2D cell cultures are not sufficient to capture the resistance caused by transporter pumps. The causes of Bortezomib resistance can be revealed by creating a complete model of the bone marrow microenvironment with 3D culture techniques, microfluidic, and organ-on-chip devices. Owing to the delivery of Bortezomib with nanoparticles, fewer side effects and targeted therapy may be possible.
2. Activation of Detoxification Systems
ABC transporters form a chemo immunity system that dynamically protects our body from the accumulation of foreign chemical agents
[35][79]. While P-gp carries unmodified neutral or positively charged hydrophobic compounds, the members of the MRP family extend the processing time of organic anions and Phase 2 metabolic products. In this sense, it is not a coincidence that GST and P-gp were found to be expressed together in a study
[36][80]. The synergy between detoxification systems and conjugating enzymes composes a very effective system for drug elimination (
Figure 1b). Endogenous compounds, lipolic substances’ biosynthesis, and excretion from cells as glutathioneed (GSH), glucoronated, and sulphated xenobiotics are of vital importance in detoxification. These substances are taken up in the cell by oxidation, glutathione, or in conjugation with alternative anionic groups while being extruded from the cell by transporter pumps. Most of the drugs are natural toxins and can also be inactivated by oxidation or conjugation. In Phase 2 reactions, the conjugation with glutathione makes them harmless and water-soluble metabolites. Only conjugation is not sufficient to remove the drug from the cell
[37][81], because such a drug is more hydrophilic. MRP transporters were shown to play a role in detoxification and glutathione-dependent drug resistance
[38][39][82,83].
Glutathione S-transferases (GSTs) conjugate electrophilic and hydrophobic compounds of endogenous or exogenous origin with glutathione. GSTs are a family of enzymes that are generally responsible for Phase 2 detoxification processes, catalysing the conversion to more easily disposable and less toxic metabolites
[40][84]. GSTs comprise various subunits with high polymorphism. Each subunit (22–29 kDa) is a dimeric protein consisting of two catalytically independent functional regions. These functional regions are hydrophilic G-regions that bind the physiological substrate GSH and H-region, which binds the hydrophobic substrates. GSH levels and expression of GST enzymes are increased by the uptake of anti-cancer agent into the tumour cell
[41][85]. Increased GSH/GST levels accelerate the metabolism of many drugs in the treatment of chemotherapy, leading to a lack of drug-targeted effects and resulting in the development of drug resistance
[42][86]. In that case, GST and MRP over expressions are in line with the synergistic effect on high-level resistance to several drugs
[7][43][35,37]. The results of Zhao et al. with MM patients showed that GSTP1 could be a biomarker for diagnosis and prognosis
[44][38]. In this sense, it is clear that 3D models, microfluidic, and organ-on-chip devices that provide full simulation of the BM microenvironment are needed to prevent bortezomib’s detoxification mechanism and excretion with MDR transporters. In addition, by directing Bortezomib with nanoparticles in a target-specific manner, extra drug use and excretion can be prevented.
3. Changes in Drug Targets
The sensitivity of multiple myeloma cells to Bortezomib is based on the fact that malignant B cells depend on protein synthesis and conversion and therefore must rely on the ubiquitin proteasome system (UPS) for processing damaged proteins
[45][87]. Myeloma cells are the most protein-secreting cells of all cell types, and these proteins, if not folded properly, are destroyed in the proteasomes (
Figure 1c). Therefore, these cells are under a constant endoplasmic reticulum stress and can easily induce unfolded protein response (UPR)
[46][88]. The efficacy of the proteasome inhibitor Bortezomib is limited by the resistance development in the disease
[47][89]. Studies with MM patient samples and cell lines have shown that Bortezomib resistance is associated with reduced IRE1/XBP1 activity and changes with the activity status of UPR
[48][39]. That resistance is associated with decreased UPS and is also an indicator of disruptions in the mechanisms of autophagy, de-ubiquitation, and chaperone proteins, which allow the cell to overcome this stress
[49][90]. In normal cellular homeostasis, autophagy appears to be a tumour suppressor, while it can direct the tumour cell survival under stress conditions
[50][91]. Autophagy initiates a survival mechanism to eliminate UPS substrates upon proteasome inhibition
[51][92]. In a study with bortezomib-resistant breast cancer cells, it was reported that increased ATF4 expression caused the induction of autophagy
[52][93]. Induction of autophagy via chaperones (Hsp70 and Hsp90) also has an effect on the survival and apoptosis of MM cells
[53][40].
The studies on molecular mechanisms underlying Bortezomib resistance have focused on developing BTZ resistant tumour cell line models
[54][94]. BTZ resistant cell line models were mutated in the β5 subunit of the proteasome, and these mutations were clustered at the S1 binding site in the PSMB5 gene
[9][59]. It was observed that different PSMB5 mutations caused different levels of BTZ resistance and continuous mutations occurred due to selective repression in long-term cultures
[55][41]. Of course, only the mutations in the β5 subunit will not be responsible for the entire resistance mechanism. Many studies emphasized that over-expression of the POMP gene plays a role in BTZ resistant cell lines
[56][95]. As known, tumour cells have a potential in directing the immunoproteasome function to get away from immune surveillance
[57][96]. It was shown that the PSMB8 gene responsible for the β5i subunit was mutated in BTZ resistant cell lines
[58][42]. These mutations caused a decrease in PSMB8 expression and chymotrypsin-like activity
[59][97]. This way, BTZ resistant cell lines could gain a high drug resistance phenotype by lowering the immunoproteasome level
[60][98]. siRNA and miRNA technologies can be used to elucidate other proteasome mutations and their functions or other mechanisms for Bortezomib resistance in MM. The use of 3D models, microfluidic, and organ-on-chip devices in combination with siRNA/miRNA technologies will greatly contribute to the examination of the Bortezomib resistance profile at the organism level.
4. Domination of Cell Cycle or Apoptosis
Cell cycle and apoptosis function in cancer cells are impaired for many different reasons (
Figure 1d). Mutations for the activation of oncogenes such as NRAS, KRAS, BRAF, and CCND1 and inhibition of tumour suppressors such as RB1, DIS3, CDKN2A, and CDKN2C are involved in the development of MM
[61][99]. P-53, known as the guardian of the genome, is a protein that has excessive mutations in cancer patients and fulfils this role by mediating the degradation of numerous cell cycle regulators and apoptotic factors (Bcl-2, p21, p27, c-Myc, cyclin A, B, D, E). When p53 mutation occurs in MM cells, the relevant signalling pathways and targets were shown to cause the development of anti-apoptosis and drug resistance
[62][63][43,46]. The overexpression of c-Myc on chromosome 8q24 is also associated with disease aggression and Bortezomib resistance
[64][44]. MM cells with MAF overexpression were resistant to Bortezomib by inhibiting apoptosis
[65][45]. All of these contribute to oncogenesis by promoting MM progression and drug resistance.
Apoptosis is an energy-dependent programmed cell death, regulated by the organism. This process plays a critical role in the maintenance of tissue homeostasis as well as the destruction of damaged or potentially dangerous cells. Chemotherapy can substantially kill tumour cells with apoptosis, while inhibition of apoptosis can make tumour cells become resistant to chemotherapy
[66][100]. Suppressing apoptosis provides an advantage to the cancer cell by reducing cell forfeit
[67][101]. MM cells induce apoptosis by changing FAS, TNF-associated ligands, or Bcl-2/Bax ratio
[63][46]. Changes in Bcl-2 and Bax regulation were observed in MM cells following Bortezomib treatment
[68][102]. Apoptosis-suppressed MM cells increase the regulation of antiapoptotic factors (Bcl-xL, Mcl-1, Bcl-2), upregulate apoptosis inhibitors, and acquire resistance to FAS, TNF ligands that induce apoptosis
[69][47]. TNF-α and FasL family member TRAIL/Apo2L reversed Bortezomib resistance in MM cells
[70][103]. MM cells express high levels of PD-L1, which helps them evade immune cells. Increased PD-L1 expression in MM cells stimulated with IFN-γ and TLR ligands escapes from cytotoxic T lymphocytes by inhibition of MyD88/TRAF6 and MEK/ERK/STAT1
[71][104]. The resistant tumour cells get rid of drug-induced apoptosis by excreting the drug from the cell, and ABC transporters cause multi-drug resistance in tumour cells not only by drug excretion but also by apoptosis and cell cycle signalling pathways
[72][105]. Furthermore, glutathione conjugates of many anti-cancer drugs regulate the stress-activated apoptosis pathway through GST isoenzymes
[73][106].
Apart from these, it was noticed that some miRNAs targeting genes that regulate cell cycle, apoptosis, survival and cell growth in MM are dysregulated
[74][107]. For example, miR-106b-25 cluster, miR-181a, miR-181b, and miR-32 together regulate p-53
[75][108]. The miR-17-92 cluster regulates Bcl-2
[76][109], miR-29b Mcl-1
[77][110], miR-21 STAT3
[78][111], and miR-125b BLIMP1
[79][112]. Furthermore, Neri et al. identified an MM miRNA signature that was critical in the development of resistance to Bortezomib
[80][48]. The genetic mechanisms responsible for the development of Bortezomib resistance in multiple myeloma can be complex
[81][113]. Therefore, 3D modelling of the factors that push MM cells to apoptosis and resistance to bortezomib, both in terms of genetics and the tumour microenvironment, might be a solution for re-sensitizing myeloma cells. Gene silencing and personalized treatment options will be possible with microfluidic and organ-on-chip devices that can be used for this purpose. Moreover, with the targeting ability of nanoparticles, specifically MM cells will be able to undergo apoptosis.
5. Distortion of Signalling Pathways
Inhibiting proteasomes with Bortezomib disrupts various cell signalling pathways, leading to apoptosis, cell cycle arrest, and supressing angiogenesis (
Figure 1e). Cancer cells can prevent drug-induced apoptosis by activating survival factors. The interaction of myeloma cells between bone marrow (BM) stromal cells and extracellular matrix (ECM) proteins is vital for ensuring the release of growth factors and cytokines. Bortezomib prevents the binding of myeloma cells to ECM proteins and BM stromal cells. The proliferation of MM cells is triggered by cytokines such as IL-6, IL-21, IGF-1, VEGF, TNF-α, SDF-1α, and the RAF/MEK/MAPK signalling cascade in the BM microenvironment
[82][114]. NF-kB activity in myeloma cells is important for maintaining the interaction with BM stromal cells, because this factor regulates the expression of IL-6, VEGF, and IGF-1, which provides the survival, development, and chemoresistance of myeloma cells around BM
[83][49]. Subsequently, JAK/STAT3 and PI3K/AKT signal cascades take place. NF-kB allows the expression of genes that protect cells from drug-induced apoptosis, thereby reducing the effectiveness of chemotherapy
[84][50]. While Bortezomib performs apoptotic function by inhibiting the canonical pathway of NF-kB, it induces the non-canonical pathway that makes myeloma cells less susceptible to Bortezomib at the same time
[85][115]. Thus, myeloma cells could develop a bortezomib-resistant NF-kB phenotype
[86][116]. Inhibition of NF-kB activation might cause DNA damage via the atypical pathway in myeloma cells and result in the actuation of multiple survival mechanisms.
BM microenvironment mediated drug resistance is defined by soluble factor (SFM-DR) and cell adhesion (CAM-DR) drug resistance mechanisms
[87][117]. MM precursor cells with a high expression of adhesion molecules are drug resistant and selected with the contribution of CAM-DR during treatment process
[88][51]. SFM-DR can be best described by IL-6 affinity, because IL-6 secretion leads to Bortezomib resistance in myeloma cells
[89][52]. Likewise, myeloma cells showed Bortezomib resistance by IL-8 released from BM stromal cells
[90][118]. In addition, MARCKS is a protein that plays an important role in cell adhesion, spreads invasion, and has recently been implicated in metastasis
[91][119]. The ability of exosomes to deliver various molecules might affect Bortezomib resistance. Indeed, it was reported that BMSC-derived exosomes could inhibit MM cell death
[92][120]. It was also found that increased RARα-2 expression contributed to drug resistance in MM CSCs
[93][121]. With the contributions of 3D culturing, microfluidic and organ-on-chip devices that recapitulate the disrupted signalling pathways and tumour microenvironment, we can understand how the processes lead to MM Bortezomib resistance function. Thus, personalized and bedside treatment will be possible, and new generation drugs and different advancing technologies will be realized.