1. Fundus Diseases and Pathological Changes
1.1. CD36 and Age-Related Macular Degeneration (AMD)
Age-related macular degeneration (AMD) is a complex eye disease that often causes irreversible blindness in the elderly. The degeneration of RPE, choroidal capillaries, and photoreceptor cell death were the main manifestations
[1][121].
Phagocytic cells are mainly divided into specialized and non-specialized types, which distinguish living cells and designated cell materials such as living cell fragments and apoptotic cells by receptors such as MerTK and CD36 expressed on the cell membrane
[2][3][122,123]. Unlike the previous two, the specialized phagocyte type is a relatively new phagocyte type that includes RPE and Sertoli cells
[2][122]. Although specialized phagocytes and non-specialized phagocytes are epithelial-derived stromal cells, they have functions such as glucose and cholesterol transport, barrier, support, and immune regulation
[4][124]. Phagocytic RPE is a polarized monolayer of epithelial cells that performs many photoreceptor health maintenance functions and participates in the absorption, transport, and degradation of photoreceptor outer segments (POS)
[5][6][11,12]. The POS located at the tip of the photoreceptor is composed of a multi-layer phospholipid bilayer, which are shed with circadian rhythm and are bound, recognized, and phagocytosed by the apical end of RPE cells with phagocytosis function in order to carry out effective metabolism to remove photooxidative wastes accumulated in the process of light transduction
[7][125]. This process occurs mainly in the morning, and each RPE cell serves about 25 POS, making it arguably the busiest macrophage in the body
[8][95]. In conclusion, RPE and photoreceptors play a synergic role in the maintenance of photoconduction homeostasis. Studies have shown that oxidative damage of RPE leads to dysfunction, which is a key component in the pathogenesis of AMD and may promote the release of extracellular vesicles (EVs) from RPE
[5][9][11,97]. If POS phagocytosis is not rhythmic, there is a late-onset cumulative phenotype characterized by visual loss and lipofuscin accumulation, which is typical of AMD
[10][96].
The receptor-mediated phagocytosis of POS by RPE is divided into two independent steps. Firstly, the integrin αvβ5 receptor initiates the signaling pathway
[10][96], and secondly, MerTK activates the POS internalization mechanism
[11][98]. Studies from rat (RPE-J) or human (ARPE-19) stable RPE cell lines found that CD36 expression is stable
[8][95]. Blocking CD36 by CD36 antibody or anionic phospholipid can partially inhibit the uptake of POS by RPE in vitro
[12][126]. CD36 as a phosphatidylserine (PtdSer/PS) receptor is sufficient to confer the ability of non-macrophage RPE to phagocytose apoptotic cells to participate in the elimination of POS
[13][127].
The secondary transmembrane protein CD36 is associated with lipid rafts in macrophages, and the finding that scavenger receptors, including CD36, localized and/or migrated in whole or part during POS phagocytosis by RPE, confirmed that RPE cells have a similar situation to macrophages
[8][95]. Some studies have shown that CD36 is involved in POS binding process but not internalization
[6][12]. Zhao et al. suggested that RPE cells have a double function of engulfing rod outer segment membranes and fibronectin, while rod outer segment membranes lack a competitive effect on fibronectin. This indicates that RPE uses different receptors in the phagocytosis of the two. Phagocytosis of rod outer segment may include CD36 and αVβ5
[14][128], and phagocytosis of fibronectin is mainly mediated by α5β1 integrin. Phagocytosis of fibronectin is mainly mediated by α5β1 integrin
[15][129]. Many studies have also shown that CD36 does not participate in the αVβ5 integrin-dependent phase of RPE phagocytosis, acts independently of αVβ5, and only participates in POS internalization
[16][17][130,131]. Thus, CD36 may function primarily as a signaling molecule. Roggia et al. upregulated peroxisome-proliferator-activated receptor γ coactivator-1α (PGC-1α) by siRNA and blocking antibodies of CD36 and MerTK, while αVβ5 integrin siRNA and FAK inhibitors inhibited PGC-1α upregulation
[6][12]. Thus, it was proved that αVβ5 integrin and FAK, not CD36, upregulated PGC-1α. Through αVβ5 integrin /FAK/PGC-1α pathway, it alleviated choroidal capillary dysfunction, lysosome accumulation, and Bruch’s membrane (BM) thickening, thereby protecting RPE.
Chang et al. used immunoprecipitation and antibody inhibition experiments to show that it is the interaction of CD81 on RPE rather than CD9, Mer tyrosine kinase, or CD36 with αVβ5 integrin that regulates the availability of αVβ5 integrin binding to POS particles and maintains metabolic stability by changing the activity of αVβ5 receptor
[18][132], participating in the first step of POS engulfing by RPE. Therefore, POS phagocytosis by RPE requires both αVβ5 and CD81, independent of CD36.
POS membrane phospholipid content of close to 25%, mainly composed of neutral phospholipid of phosphatidyl choline (PC) and phosphatidyl ethanolamine (PE), accounted for about 80%. Negatively charged PS and phosphatidylinositol (PI) accounted for 13% and 7%, respectively
[12][126]. In pathological cases, abnormal external exposure of PS and PI may be specifically recognized and phagocytosed by circulating monocytes or macrophages
[19][133].
Oxidized phosphatidylcholines (oxPCs), a high-affinity ligand for CD36 (oxPC
CD36), selectively inhibits ox-LDL binding to CD36 transfected cells. OxPC
CD36 produced by oxidation is widely present in atherosclerotic plaques
[20][134]. Sun et al. found that oxidative stress induced by intense light in the dark-adapted rat retina “oxidized” phosphatidylcholine in the outer segment of photoreceptors, producing a new structure-specific oxidized phosphatidylcholine molecule oxPC
CD36 from 1-palmitoyl-2-linoleyl-
sn-glycerol-3-phosphatidylcholine (PLPC), 1-palmitoic-2-arachidonyl-sn-glycerol-3-phosphatidylcholine (PAPC), and the docosahexaenoate ester of 2-lysophosphatidylcholine (DHA-PC)
[19][133]. CD36 knockdown mice were used to demonstrate that RPE-mediated POS internalization was achieved through a specific interaction between CD36 and oxPC
CD36 [19][133]. Experiments by Ryeom and Sparrow et al. showed that it was PS, and PI liposomes but not PE that competitively bound RPE with purified POS and was internalized. However, no binding of RPE to PS or PI was found in the RPE of mutant rats without CD36 expression. Therefore, oxPCs, PS, and PI may be physiological signals for CD36 to recognize POS on RPE
[12][126].
Using human retinal epithelial cells (ARPE19) in vitro, Gordiyenko et al. revealed that RPE cells internalize ox-LDL via CD36
[21][135]. CD36 of RPE cells in AMD may internalize oxidized form of cholesterol through LDL, leading to subretinal lipid deposition and accumulation of ox-LDL at the basal side of RPE or BM level, causing lipid metabolism disorders and producing the drusen characteristic of AMD. This causative factor in the membrane region of RPE-Bruch is similar to the cytotoxicity of ox-LDL accumulation in the atherosclerotic machinery
[21][135]. Studies have shown that the phagocytic capacity of CD36 seems to be enhanced under the conditions of lipid stress or oxidative POS in AMD eyes
[22][136]. Studies on macrophages have also shown that macrophages have a higher binding capacity to oxidative PtdSer
[23][137]. The conclusions of these two studies are consistent.
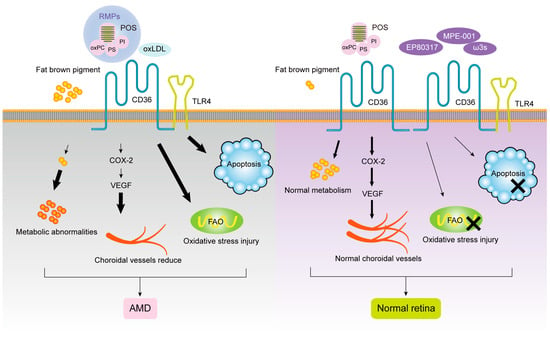
Houssier et al. suggested that CD36 deficiency downregulates retinal POS-induced proangiogenic cyclooxygenase (COX)-2 and vascular endothelial growth factor (VEGF) expression, resulting in choroidal capillary rarefacialization and photoreceptor and choroidal degeneration, leading to dry AMD
[24][66]. As an important participant in POS phagocytosis by RPE, CD36 receptor density affects phagocytosis kinetics, which can be used as one of the criteria for the judgment of cell phagocytosis. Westenskow et al. demonstrated that RPE from the induced pluripotent stem (IPS-RPE) had a good phagocytosis function for POS by flow cytometry detection of αVβ5 integrin, CD36, MerTK receptor expression density, and binding and internalization kinetics at different differentiation stages of RPE. It may be a good substitute for diseased RPE
[16][130]. The ability of CD36 to mediate anti-angiogenesis of TSP-1 suggests that CD36 dysfunction may cause neovascularization in addition to abnormal phagocytosis
[25][138]. Kondo et al. used TaqMan genotyping to detect 19 single-nucleotide polymorphisms in CD36 in 109 neovascular AMD and 182 unrelated control subjects, finding that two variants in CD36, rs3173798 and rs3211883, were associated with neovascular AMD. These results suggest that CD36 may be a new candidate susceptibility gene for neovascular AMD
[26][139]. Honda et al. used the TaqMan probe method to genotype 19 single-nucleotide polymorphisms of CD36 in 73 polypoidal choroidal vasculopathy (PCV) patients who responded to photodynamic therapy (PDT) treatment and 64 PCV patients who did not respond to treatment. The results showed that the CD36 rs3173798 variant may be associated with the visual prognosis of PCV patients with PDT
[27][140]. As PCV is a subtype of AMD
[28][141], this study further suggested that CD36 may provide genetic information for the development of AMD.
Studies have found that RPE cells are involved in oxidation-induced release and uptake of EVs, which can in turn act as a factor to accelerate the oxidative damage of AMD. Aged RPE cells release more RPE-cell-derived microparticles (RMPs), which can accelerate the senescence of RPE cells and interrupt the phagocytosis activity. Because blocking CD36 effectively attenuates the uptake of RMPs by RPE cells, it is speculated that CD36 on RPE accelerates the formation of AMD by participating in the uptake of RMPs
[9][97].
EP80317, a novel ligand of selective CD36, is a derivative of growth-hormone-releasing peptide-6 (GHRP-6), which lacks growth-promoting activity due to the presence of lysine 3 and is considered to have potential anti-atherogenic activity
[29][142]. Picard et al. found significant AMD represented by increased thickness of BM in
ApoE−/− high-fat cholesterol-fed mice by electron microscopy. Later studies found that
CD36 and
ApoE double-knockout normal-diet fed mice also showed age-related subretinal oxLDL accumulation and BM thickening, while
ApoE−/− high-fat diet mice injected with CD36 agonist EP80317 through the tail vein had the above pathological features significantly reduced and they retained some photoreceptor function. The mechanism may be that EP80317 promotes the clearance of oxidized lipids in BM and maintains photoreceptor function by increasing the expression of CD36 protein on RPE. Therefore, CD36 may be a promising therapeutic target for AMD
[30][143]. Nitrogen impurity peptide MPE-001 (His-D-TRP-Ala-acetyl-d-Phys-NH2) is an amino derivative of GHRP-6 with high CD36 binding affinity
[31][144]. Dorion et al. elucidated the role of MPE-001 as a CD36 ligand in the cytoprotective mechanism of RPE by applying the MPE-001 to a model of AMD oxidative stress developed from sodium iodate and RPE cell lines
[5][11]. Kindzelskii et al. found that CD36 arrived at the POS–RPE cell interface, followed by TLR4 aggregation within 2 min, followed by metabolic and calcium signaling, suggesting that TLRs after CD36 are involved in RPE transmembrane metabolism, calcium signaling, and ROM release in RPE uptake of POS
[32][16]. Diet plays a role as a regulator of fatty acid structure in the neurosensory retina. Promoting the intake of ω3 long-chain polyunsaturated fatty acids and decreasing the linoleic acid diet can upregulate the expression of the
CD36 gene involved in lipid transport and angiogenesis in the neurosensory retina of rats, as well as change the fatty acid profile. This is consistent with the findings of epidemiological studies related to AMD, but no effect on retinal function was observed. Therefore, the optimization of diet structure may provide a new therapeutic direction for the prevention of AMD retinal function damage through CD36. In the pathogenesis of AMD, given the important function of CD36 in POS phagocytosis by RPE, recovery/activation of CD36 expression by specific recognition of ligands may provide new ideas for the treatment of AMD. The role of CD36 ligands and signal transduction pathways in the progression of AMD is shown in
Figure 12.
Figure 12.
The roles of CD36 in AMD.
1.2. CD36 and Diabetic Retinopathy (DR)
Diabetic retinopathy (DR) is a vascular abnormality including basement membrane thickening, pericyte loss, microaneurysm formation, and capillary leakage
[33][145]. Chronic low-grade inflammation caused by abnormal expression of proinflammatory cytokines in retinal cells plays an important role in the development of DR; can adjust abnormal biological and biochemical processes of pericytes, endothelial cells, and microglia; impairs cell proliferation, endothelial cell tight junctions, and other cell function; and lead to apoptosis, ultimately causing vision loss
[34][146]. Leukocyte adhesion to retinal vessels and proinflammatory cytokine release are two important markers of early vascular inflammation in DR
[35][101].
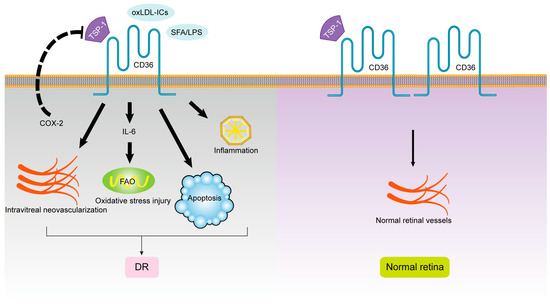
COX-2 is an immediate early gene product induced by inflammatory cytokines, mitogens, and endotoxins, leading to an increase in prostaglandins during inflammation. Sennlaub et al. found that COX-2-inhibitor-induced upregulation of anti-angiogenic factor TSP-1 and CD36 receptors in endothelial cells prevented intravitreal neovascularization. Prostaglandin E2 reverses the effect of COX-2 inhibitors on TSP-1 and CD36 and aggravates the formation of intravitreal neovascularization. Moreover, wb results suggested that this effect might be independent of VEGF. Therefore, COX-2 may play an important role in ischemic proliferative retinal diseases such as diabetic retinopathy by inhibiting CD36
[36][147].
Modification of LDL may make it immunogenic, leading to the formation of LDL immune complexes (LDL-ICS)
[37][148]. Elevated circulating ox-LDL-ICS levels were found to predict the risk of severe nonproliferative DR and proliferative DR in type 1 diabetes mellitus
[38][149]. Plasma levels of malondialdehyde modified apolipoprotein B-100 antibody are positively associated with the severity of DR in type 2 diabetes mellitus, and the importance of ox-LDL-ICS is further emphasized
[39][102]. Ox-LDL-ICS is involved in the induction of retinal oxidative stress, endoplasmic reticulum stress, and apoptosis; increases the secretion of inflammatory cytokines; and reduces the secretion of the key anti-angiogenic factor pigment epithelium-derived factor, having a toxic effect on retinal capillary pericytes. After blocking CD36, the oxidative stress level and ER-stress-mediated apoptosis of retinal pericytes were attenuated
[40][150]. Thus, CD36 mediates the interaction of pericytes with ox-LDL-ICS, but no similar effect was found in mesangial cells
[41][151].
Free fatty acids, especially saturated fatty acids (SFA), were found to upregulate the expression of proinflammatory cytokines
[42][152]. Clinical studies have shown a correlation between serum levels of saturated fatty acids and the severity of DR
[43][153], with palmitate being the most abundant SFA in humans
[44][154]. Lu et al. found that human retinal microvascular endothelial cells (HRMVECs) expressed CD36 in vitro. It is also involved in the upregulation of IL-6 by lipopolysaccharide (LPS), palmitate, or LPS+ palmitate, which may trigger inflammatory signals such as the JNK cascade
[45][14]. Therefore, it is speculated that HRMVECs participate in palmitate-induced signal activation and subsequent gene expression through CD36, causing DR
[46][155].
In conclusion, lowering blood lipids, reducing oxidative stress and ox-LDL production, and antagonizing CD36 receptors may effectively prevent diabetic retinopathy. The role of CD36 ligands and signal transduction pathways in the progression of DR is shown in Figure 23.
Figure 23.
The roles of CD36 in DR.
1.3. CD36 and Glaucoma
Glaucoma, the world’s first irreversible cause of blindness, is characterized by progressive RGC loss in retinal ganglion cells. In glaucoma and other neurodegenerative diseases, such as Alzheimer’s disease, microglia, as resident immune cells in the central nervous system, can mediate neuroinflammation; recognize and bind Aβ through membrane receptors, especially CD36; and exacerbate pathological progress
[47][48][156,157].
Simons et al. found that Aβ mediates retinal microglia inflammation through CD36 activation, induced different patterns of RGC degeneration loss, and increases glial cell proliferation and activation, promoting the production of reactive oxygen species (ROS) and the secretion of proinflammatory cytokines including IL-1β and TNF-α, thereby generating intracellular signaling cascades, which may be the mechanism of retinal CD36-mediated RGC injury
[49][104]. However, mice lacking CD36 receptors showed significantly reduced Aβ-mediated retinal RGC damage
[50][158]. Therefore, inhibition of CD36-mediated amyloid Aβ accumulation in the retina can effectively prevent RGC loss, which may provide A new idea for the treatment of glaucoma.
1.4. CD36 and Retinal Neovascularization
In mice born blind, vision after birth depends on the development of retinal blood vessels, and retinal hypoxia leads to neovascularization.
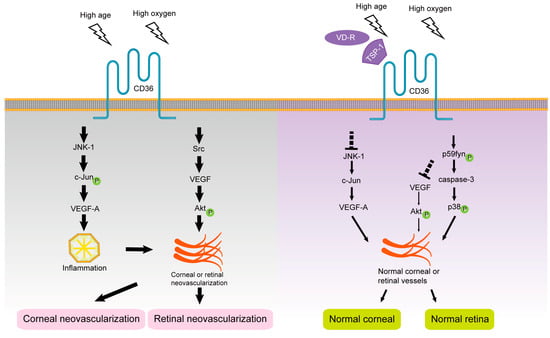
TSP-1 has multiple domains and interacts with many cell surface receptors in vertebrates
[51][119]. As a stromal cell calcium-binding protein, TSP-1 regulates cytoskeletal organization, adhesion, migration, and apoptosis; limits vessel density in normal tissues; and blocks pathological angiogenesis and development, making it a naturally occurring angiogenesis inhibitor
[52][159]. Jiménez et al. showed that as a receptor for TSP-1, CD36 is involved in the angiogenesis blocking effect of TSP-1 and activates subsequent downstream signaling pathways including p38 MAPKs, p59Fyn, and caspase-3-like proteases. Co-immunoprecipitation assay suggested that Fyn was a mediator of the negative function of TSP-1/CD36, while Src provided a positive signaling pathway promoting microvascular survival
[53][120]. Tian et al.’s study on rhesus-macaque-derived choroid-retinal endothelial (RF/6A) cells found that VR-10 peptide (Val-Thr-Cys-Gly-Val-Ile-Thr-Arg-Ile-Arg) located at the anti-neovascularization site of TSP-1 could interact with its receptor CD36 to regulate the generation of choroidal anti-neovascularization
[54][160].
Hypoxic conditions increase the mRNA stability and expression level of VEGF, which explains the downregulation of endogenous VEGF-A expression in mouse pups under hyperoxia, leading to apoptosis of immature vascular endothelial cells that are not encapsulated by pericytes in vivo
[55][161]. Chu et al. found that the binding of TSP-1 to CD36 promotes the binding of Src-homology-2-domain-containing protein tyrosine phosphatase-1 to CD36-vascular endothelial growth factor receptor 2 (CD36-VEGFR2) complexes in microvascular endothelial cells, attenuating VEGF signaling, dephosphorylating VEGFR2, and inhibiting angiogenesis
[56][162]. Sun et al. used co-immunoprecipitation and other experiments to show that the presence and absence of TSP-1 recruit Fyn or Src to the CD36 membrane domain, respectively, to regulate microvascular remodeling in the developing retina by antagonizing or promoting VEGF-driven Akt signaling phosphorylation
[57][103]. These results suggest that TSP-1 may inhibit VEGF-involved retinal neovascularization through the TSP-1/CD36/Fyn pathway, and thus CD36 may be a potential therapeutic target for retinal neovascularization. The role of CD36 ligands and signal transduction pathways in the progression of retinal neovascularization are shown in
Figure 34.
Figure 34.
The roles of CD36 in intraocular neovascularization.
1.5. CD36 and Subretinal Inflammation
Inflammation is an important component of retinal degenerative diseases. Mononuclear phagocytic cells (MP; by monocytes, of macrophages and microglia cells) in the subretinal space lead to activation and aggregation, with proinflammatory and potential neurotoxicity, causing degeneration of RPE and photoreceptors and eventually causing retinal degenerative diseases such as diabetic retinopathy and age-related macular degeneration
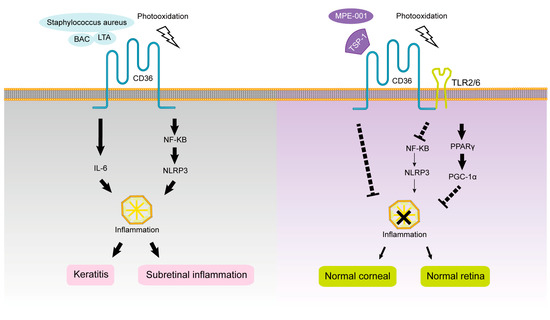
[58][59][163,164]. Abe et al. showed that CD36 is co-expressed on the surface of MP with a TLR2/6 heterodimer combination, participates in the clearance of various debris, maintains TLR2/6 signaling induced by diacylglycerol, and regulates TLR2-dependent macrophage-driven inflammatory response
[60][165]. Alterations in the metabolic rate of MP, such as inhibition of glycolysis or oxidative phosphorylation, alter the activation of M1 or M2 in different inflammatory profiles, respectively
[61][166].
Mellal et al. found that CD36-deficient mice have less subretinal MP accumulation and inflammatory cytokine infiltration and preserve the integrity and function of photoreceptor structure, which is involved in the development and treatment of degenerative retinal diseases. The CD36 selective nitrogen impurity peptide ligand MPE-001 in wild-type mice can specifically modulate CD36–TLR2 interactions to modulate the inflammatory profile and subsequent neurotoxicity of MP. MPE-001 also causes the metabolic pathway of M1-type MP to change from a glycolytic state to a state favoring oxygen consumption. Therefore, MPE-001 is expected to target the CD36 receptor and reduce chronic retinal inflammation driven by MP. MPE-001 inhibits some CD36 signaling pathways, such as NF-κB and NLRP3 inflammasome activation, and attenuates the inflammasome cascade and alters metabolic rate to increase oxygen consumption by activating other signaling pathways, such as PPARγ/PGC1α
[62][105]. Lavalette et al. used
Cx3cr1−/− and
Cx3cr1−/−CD36−/− mice and fluorescent staining, finding that in
Cx3cr1−/− mice with light-induced subretinal inflammation, IL-6 enables mononuclear phagocytes to survive and accumulate in the subretinal under immunosuppressive conditions, causing photoreceptor cell degeneration, and this process depends on CD36. Studies suggest that CD36 may be involved in the formation of subretinal sterile inflammation
[63][167]. The role of CD36 ligands and signal transduction pathways in the progression of intraocular inflammation are shown in
Figure 45.
Figure 45.
The roles of CD36 in intraocular inflammation.
2. Ocular Surface Diseases and Pathological Changes
2.1. CD36 and Corneal Neovascularization (CNV)
The angiogenic privilege of the cornea refers to the phenomenon that the cornea is normally devoid of blood vessels and actively maintains this avascular state. Although the occurrence of neovascularization is beneficial to the reconstruction of damaged tissues, good optical clarity and an unobstructed light path are very important for the maintenance of good visual acuity for some tissues or organs lacking blood vessels, such as the cornea
[64][168]. Corneal neovascularization (CNV) extending centrally from the limbal vascular plexus blocks the passage of light, resulting in corneal opacity, irreparable damage to the structure and function of the tissue, and even the final loss of vision, and therefore blocking CNV in the cornea is very important.
The results of the A gene chip related to CNV by Ren et al. showed that serum amyloid A (SAA) is mainly produced by hepatocytes
[65][106]. SAA and formyl peptide receptor 2 (Fpr2), which are closely related to inflammation, were upregulated, suggesting that they may be closely related to CNV, but CD36 was not involved in this pathway
[66][107]. Mwaikambo et al. showed that the corneal epithelium constitutively expresses CD36, which acts as an endogenous antiangiogenic receptor binding to a variety of ligands including TSP-1, ox-LDL, and apoptotic cells
[67][169]. The corneas of CD36-deficient mice showed no significant changes at 4 weeks of age but showed significant age-dependent CNV in the central cornea with corneal scar formation and corneal stromal inflammatory cell infiltration at 52 weeks of age, suggesting that CD36 can effectively inhibit inflammatory corneal neovascularization. It is speculated that this alteration is secondary to increased pathological changes such as age-dependent scar formation and chronic inflammation rather than the result of
CD36 gene knockdown alone. Because the ability of the cornea to respond to environmental or inflammatory insults declines in aged mice, the protective effect provided by CD36 becomes more important with aging
[68][13]. This effect was associated with reduced corneal transcription of the antiangiogenic factor TSP-1 and increased mRNA levels of VEGF-A, JNK-1, and c-Jun, with activation of JNK-1 and subsequent c-Jun phosphorylation required for the angiogenic effects of VEGF-A. In addition, macrophages have been shown to provide essential VEGF for inflammatory CNV.
The role of CD36 ligands and signal transduction pathways in the progression of corneal neovascularization are shown in Figure 34.
2.2. CD36 and Keratitis
In the cornea, epithelial cells must migrate as an intact sheet to maintain barrier function, which involves cell-to-matrix and cell-to-cell adhesion. Epithelial cells that are detached from the basement membrane undergo apoptosis, and therefore adhesion is essential for the survival of adherent cells on the basement membrane
[69][108]. Due to the barrier function of the cornea, mice are highly resistant to infection by staphylococci, the normal bacterial flora on the ocular surface, and thus spontaneous bacterial keratitis usually does not occur
[70][170]. However, the damage to any one of the multiple barriers that prevent bacterial adhesion and invasion can increase bacterial susceptibility
[69][108]. CD36 is expressed in corneal and limbal epithelial cells and its primary function appears to be to maintain a physical barrier that prevents bacterial binding and is a key component of resistance to infection
[71][171]. Yoon et al. also found that corneal fibroblasts are macrophage-derived fibroblasts, which may have the activity of macrophage adhesion and phagocytosis as well as the removal of cell debris
[72][69]. Klocke et al. performed Lc-biotin staining of mildly deficient
CD36−/− corneas and showed that the basal epithelium was detached from the basement membrane with loss of tight junctions, suggesting that cell adhesion defects may lead to the formation of mild corneal defects, signifying that CD36 is involved in maintaining the structural integrity of the corneal epithelial cells and is age related. Human corneal and conjunctival epithelial cell lines were exposed to benzalkonium chloride for 5 or 15 min as proinflammatory or proapoptotic stimuli, respectively. The established ocular surface inflammation model showed downregulation of CD36 mRNA expression, suggesting that CD36 may be involved in the inflammatory and apoptotic processes of both epithelial cells
[69][108].
In addition to maintaining the cellular integrity of the corneal epithelium through its adhesion function and acting as a physical barrier to bacterial binding, CD36 also serves to maintain the avascular nature of the cornea and prevent CNV. In a mouse model of inflammatory CNV, CD36 inhibited CNV through indirect inhibition of macrophage-derived VEGF-A and direct inhibition of vascular growth expression
[69][108]. Age-dependent CNV with increased expression of VEGF and inflammation has been found in
CD36−/− mice. Before neovascularization in
CD36−/− mice, a mild corneal defect occurs, characterized by loss of epithelial tight junctions, breakdown of the mucin layer, and mild macrophage infiltration in the matrix underlying the epithelial defect occurred before CNV
[71][171]. Thus, neovascularization may occur secondary to corneal epithelial defects and subsequent association with normal microbiota in
CD36−/− mice, rather than spontaneously.
Klocke et al. found that the protective innate immune responses and the ability of macrophages to phagocytose bacteria were significantly reduced by intravenous Staphylococcus aureus in
CD36−/− mice. However, TSP1
−/− and TLR2
−/− mice did not develop spontaneous keratitis, and a new function of CD36 in maintaining the corneal epithelial barrier against infection was hypothesized to be independent of TSP-1 and TLR2, which is somewhat different from the results of Laura et al
[69][108]. The latter study showed that exogenous TSP-1 treatment increased CD36 protein and mRNA levels in both human corneal and conjunctival epithelial cell lines, inducing inflammatory and apoptosis-related changes. However, the effect on CD36 was different. CD36 protein expression in corneal epithelial cells increased immediately after TSP-1 treatment. However, after 24 h of TSP-1 treatment, the CD36 protein expression level was not significantly different in corneal epithelial cells but increased in conjunctival epithelial cells, which may be related to the high level of secretion of TSP-1 in the basal corneal epithelium. Therefore, whether TSP-1 is involved in the development of bacterial keratitis requires further investigation
[73][172].
The role of CD36 ligands and signal transduction pathways in the progression of keratitis are shown in Figure 45.