Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Claudio Tirelli and Version 3 by Dean Liu.
Idiopathic pulmonary fibrosis (IPF) is a rare, chronic, fibrosing and progressive disease limited to the lung with a largely unknown etiology and a poor prognosis. IPF is associated with a characteristically defined radiological and/or histological usual interstitial pneumonia (UIP) pattern.
- idiopathic pulmonary fibrosis
- familial pulmonary fibrosis
- genetic
- epigenetic
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a rare, chronic, fibrosing and progressive disease limited to the lung with a largely unknown etiology and a poor prognosis. IPF is associated with a characteristically defined radiological and/or histological usual interstitial pneumonia (UIP) pattern. IPF is characterized by the progressive replacement of lung tissue with fibrotic-scar tissue due to the excessive release of collagen by mesenchymal cells, the myofibroblasts. This process changes the architecture and function of the organ over time and, in the absence of antifibrotic therapy, may lead to death within 3–5 years after diagnosis. IPF is a disease of the elderly, with a mean age of onset of 65 years [1].
IPF is characterized by the presence of progressively worsening dyspnea. Cough and sputum are frequently associated symptoms, especially in more severe patients.
According to international guidelines, the diagnosis requires the exclusion of secondary causes of pulmonary fibrosis (i.e., connective tissue diseases) and the execution of a high-resolution computed tomography (HRCT) of the chest. Finding a definite usual interstitial pneumonia (UIP) radiological pattern is sufficient to diagnose IPF after the exclusion of any other causes of lung disease. Patients who have a probable UIP pattern on HRCT can receive a diagnosis of IPF after a multidisciplinary discussion (MDD) by at least a pulmonologist, a radiologist and a pathologist. Lung biopsy is today adopted only in limited complex cases, particularly when an IPF diagnosis is strongly suspected but the radiological pattern is indeterminate for UIP or suggestive for other interstitial lung diseases.
Bronchoalveolar lavage (BAL) is a bronchoscopic sampling procedure useful in the diagnostic work-up of idiopathic pulmonary fibrosis that helps rule out other fibrosing interstitial diseases that can enter into differential diagnosis with IPF. Although there are no specific cellular patterns for IPF, BAL could help in the diagnosis of other interstitial lung diseases (ILD) such as sarcoidosis, eosinophilic pneumonia, chronic extrinsic allergic alveolitis or nonspecific interstitial pneumonitis [2][3][2,3].
To date, no pharmacological therapy can stop or reverse the fibrosing process. Two drugs, nintedanib and pirfenidone, have been approved for the treatment of the disease, since in clinical trials and observational studies they have been shown to slow down the progression of the disease, without, however, being able to stop or cure it [4][5][4,5]. Several clinical trials are underway to expand the spectrum of antifibrotic drugs, but, to date, the only curative intervention remains lung transplantation, a therapeutic option applicable to a very small group of IPF patients respecting stringent selection criteria and with numerous possible complications.
Despite several advances in understanding the pathogenetic mechanisms of the disease, the origin of IPF remains largely unknown. The main risk factors include cigarette smoking, lung infections (especially viral, and, to a lesser extent, bacterial infections), exposure to pneumotoxic inhalants, gastroesophageal reflux, elderly age, diabetes mellitus and genetic predisposition [6].
Intriguingly, forms of familial pulmonary fibrosis (FPF) have long been known and linked to mutations in specific genes (Table 1). FPF is defined when the disease is diagnosed in the same family with at least 2 relatives developing pulmonary fibrosis. Altogether, these forms represent up to 20% of cases of pulmonary fibrosis [7].
Table 1. Causative genes for FPF. The relative proteins, OMIM identifier, phenotype and type of inheritance are described.
| Gene | Protein | Disease (OMIM) | Phenotype | Inheritance |
|---|---|---|---|---|
| TERT | Telomerase reverse transcriptase | PFBMFT1 (614742) | Pulmonary fibrosis and/or bone marrow failure, telomere-related | AD |
| TERC | Telomerase RNA component | PFBMFT2 (614743) | Pulmonary fibrosis and/or bone marrow failure, telomere-related | AD |
| RTEL1 | Regulator Of Telomere Elongation Helicase 1 | PFBMFT3 (616373) | Pulmonary fibrosis, adult onset | AD |
| PARN | Poly(A)-Specific Ribonuclease | PFBMFT4 (616371) | Pulmonary fibrosis, adult onset | AD |
| ZCCHC8 | Zinc Finger CCHC-Type Containing 8 | PFBMFT5 (618674) | Pulmonary fibrosis and/or bone marrow failure, adult onset | AD |
| RPA1 |
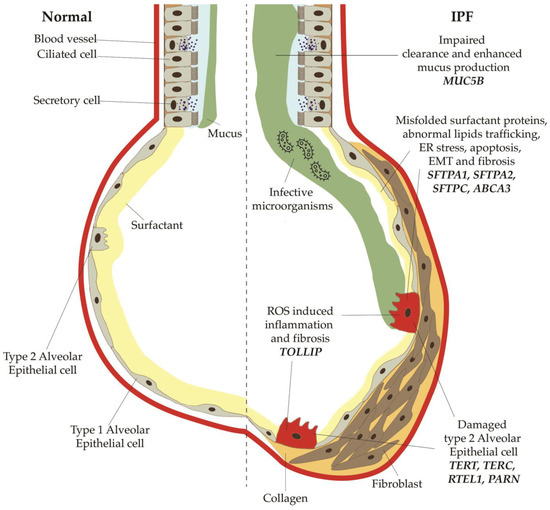
Figure 2. Main profibrotic mechanisms secondary to mutations or polymorphisms in the genes for telomerases, surfactant proteins, mucin 5B. Mutations in TERT, TERC, PARN and RTEL1 decrease the activity of telomerase, thus resulting in augmented shortening of telomeres. SFTPC, SFTPA1, SFTPA2, ABCA3 normally modulate and stabilize the alveolar surfactant tension; when altered, they can induce an increased stress of the endoplasmic reticulum, which finally lead to epithelial-mesenchymal transitions and apoptosis of type 2 alveolar cells. Polymorphisms of MUC5B, a regulatory mucin, are responsible of mucociliary disfunction, with impaired clearance and enhanced mucus production, which predisposes to bacterial overgrowth and infections.
Table 2. Genes described in idiopathic pulmonary fibrosis (IPF) and familial pulmonary fibrosis (FPF) and their profibrotic mechanisms.
| Gene 1 | Main Function | Profibrotic Mechanism | |||||
|---|---|---|---|---|---|---|---|
| TERT | Telomerase | Decreased activity of telomerase | |||||
| TERC | Reverse Transcription in Telomerase | Decreased activity of telomerase | |||||
| PARN | Stability of mRNA in Telomerase | Shortening of telomeres | |||||
| RTEL1 | DNA helicase in Telomerase | Shortening of telomeres | |||||
| SFTPA1 | Modulate surface tension in the alveoli | Increased ER stress | |||||
| Replication Protein A1 | PFBMFT6 (619767) | Pulmonary fibrosis and/or bone marrow failure and skin abnormalities | AD | ||||
| SFTPA2 | Modulate innate and adaptive immunity | Increased ER stress | SFTPA2 | Surfactant Protein A2 | ILD2 (178500) | Pulmonary fibrosis, interstitial pneumonia, lung cancer | AD |
| FAM111B | FAM111 Trypsin Like Peptidase B | POIKTMP (615704) | Poikiloderma, hereditary fibrosing, with tendon contractures, myopathy, and pulmonary fibrosis | AD |
Genes associated with FPF include those encoding for telomerases, responsible for the regulation of telomeres length and therefore involved in cellular aging (TERT, TERC, PARN, RTEL1) [8][9][10][8,9,10]. Two of the telomerase-related forms can also be characterized by bone marrow failure (PFBMFT1 and PFBMFT2 due to TERC and TERT mutations, respectively). The involvement of bone marrow function in the pulmonary fibrosis (PF) phenotype is also reported for patients with ZCCHC8 and RPA1 variants. The familiar forms of PF include ILD2 (OMIM #178500), a disease related to SFTPA2 mutations and characterized by pulmonary fibrosis, interstitial pneumonia and lung cancer.
Finally, pulmonary fibrosis can also be part of the POIKTMP syndrome (poikiloderma with tendon contractures, myopathy and pulmonary fibrosis). FPF can manifest either as IPF, but also as connective tissue disease-interstitial lung disease (CTD-ILD) or unclassifiable interstitial lung disease (U-ILD) [7][11][7,11].
On the other hand, there is little evidence of the possible role of genetics in the etiology of sporadic IPF, although mutations in FPF-causing genes are also found in about 10–20% of sporadic IPF cases.
Even if the genetic predisposition to PF is known, the molecular diagnosis of familial cases is probably underestimated, and this could be related to the incomplete penetrance of the disease or the lack of information from the patient’s pedigree. In order to recognize the familial cases, it is important to identify the risk in family members not only for PF but also for possible related conditions, mainly lung cancer and bone marrow failures. In addition to the Mendelian forms, several studies support the existence of a genetic predisposition to pulmonary fibrosis, which is expressed as a complex trait in which low-penetrance genes and environmental factors play a role. In particular, the involvement, even in sporadic forms, of genes related to the production of surfactant proteins, which would alter the functionality of the pulmonary alveoli, or of genes responsible for an increased chronic secretion of mucus, is highlighted [12].
Finally, epigenetic alterations seem to play a role in fibrogenesis. Several studies consider cellular aging as an endogenous causative factor of IPF, which favors the development of genomic instability, the establishment of epigenetic alterations, the shortening of telomeres and cellular senescence, thus laying the foundations for the development of fibrosis [13].
Overall, heterogeneous genetic variants could contribute to the development of an altered broncho-pulmonary tissue, which would become more susceptible to recurrent micro-lesions from various potential environmental factors.
2. The Genetic Basis of Idiopathic Pulmonary Fibrosis and Familial Pulmonary Fibrosis
PF can have a strong genetic component in 10–20% of cases, as previously described, or gene variants can modify the risk so that it is affected together with the influence of the environment. In addition, genetic factors might also influence the onset of the disease [14] (Figure 1).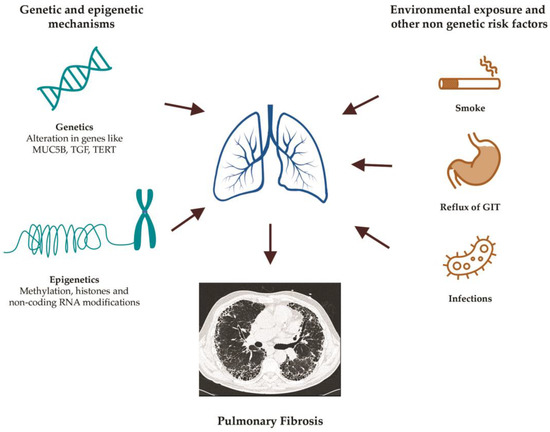
Figure 1. Main risk factors for the development of pulmonary fibrosis.
| SFTPC | ||
| Stabilize the surfactant fluid | ||
| Increased ER stress | ||
| ABCA3 | Lipid transport across membranes | Increased ER stress and apoptosis |
| MUC5B | Mucin 5B production | Muco-ciliary disfunction |
| TOLLIP | Inhibitory adaptor protein within TLR | Decreased protection against ROS |
1 TERT: telomerase reverse transcriptase; TERC: telomerase RNA component; PARN: poly(A)-specific RNase; RTEL1: regulator of telomere elongation helicase-1; SFTPA1: surfactant protein A1; SFTPA2: surfactant protein A2; SFTPC: surfactant protein C; ABCA3: ATP-binding cassette subfamily A member 3; MUC5B: mucin 5B; TOLLIP: Toll-interacting protein; TLR: Toll-like receptors; ER: endoplasmic reticulum; ROS: reactive oxygen species.
All the reported causative genes for PF show an autosomal dominant pattern of transmission, leading to a 50% chance of transmitting the pathogenetic variant to the offspring. However, the expression of the phenotype is influenced not only by incomplete penetrance, intrafamilial clinical variability and environmental factors but also by the pathomechanism related to the specific gene involved. For example, for genes involved in telomere maintenance, it is reported that the phenomenon of anticipation, is possibly favored by epigenetic mechanisms triggering telomere shortening [18].
Moreover, polymorphisms of MUC5B, a regulatory mucin, are responsible for mucociliary disfunction with impaired clearance and enhanced mucus production, which predisposes to bacterial overgrowth and infections.