The generation of phenotypic diversity is fundamental for the environmental adaptation and evolution of populations. It can be produced by genetic and epigenetic mechanisms. The genetic mechanisms include mutation, recombination, drift and gene flow and generate phenotypic diversity by changing the DNA sequence or their relative distribution in populations. Epigenetic mechanisms produce phenotypic diversity by differential expression of the same DNA without changing the DNA sequence and different processing of the gene products. They include DNA methylation, histone modifications, non-coding RNAs, Polycomb/Trithorax group proteins, chemical mRNA modifications and mRNA editing. In sexually reproducing organisms, genetic mechanisms probably play a predominant role in the production of phenotypic diversity. Epigenetic mechanisms are effective in all organisms but are particularly important in asexual reproducers, where they generate phenotypic variation and individuality despite genetic identity. Further main benefiters of the epigenetic contribution to phenotypic diversity are sessile taxa and species with long generation times.
- Phenotypic diversity
- Ecology
- Environmental adaptation
- Epimutation
- Evolution
- Genetic mechanisms
- Epigenetic mechanisms
- Individuality
1. Introduction
Phenotypic diversity in populations is fundamental for coping with environmental challenges, adaptation to new environments, and evolution. Phenotypic diversity can be generated either by genetic or epigenetic mechanisms. Both are ubiquitous in animals, plants, fungi, protists and bacteria, but their relative contribution to phenotypic diversity varies largeenormously depending on mode of reproduction, life style and life history.
2. Generation of Phenotypic DiveVarsityiation by Genetic Mechanisms
3. Generation of Phenotypic DiveVarsityiation by Epigenetic Mechanisms
4. Stochastic and Environmentally-Induced Epimutations asnd Triggers of Non-GeneticRelated Phenotypic Change
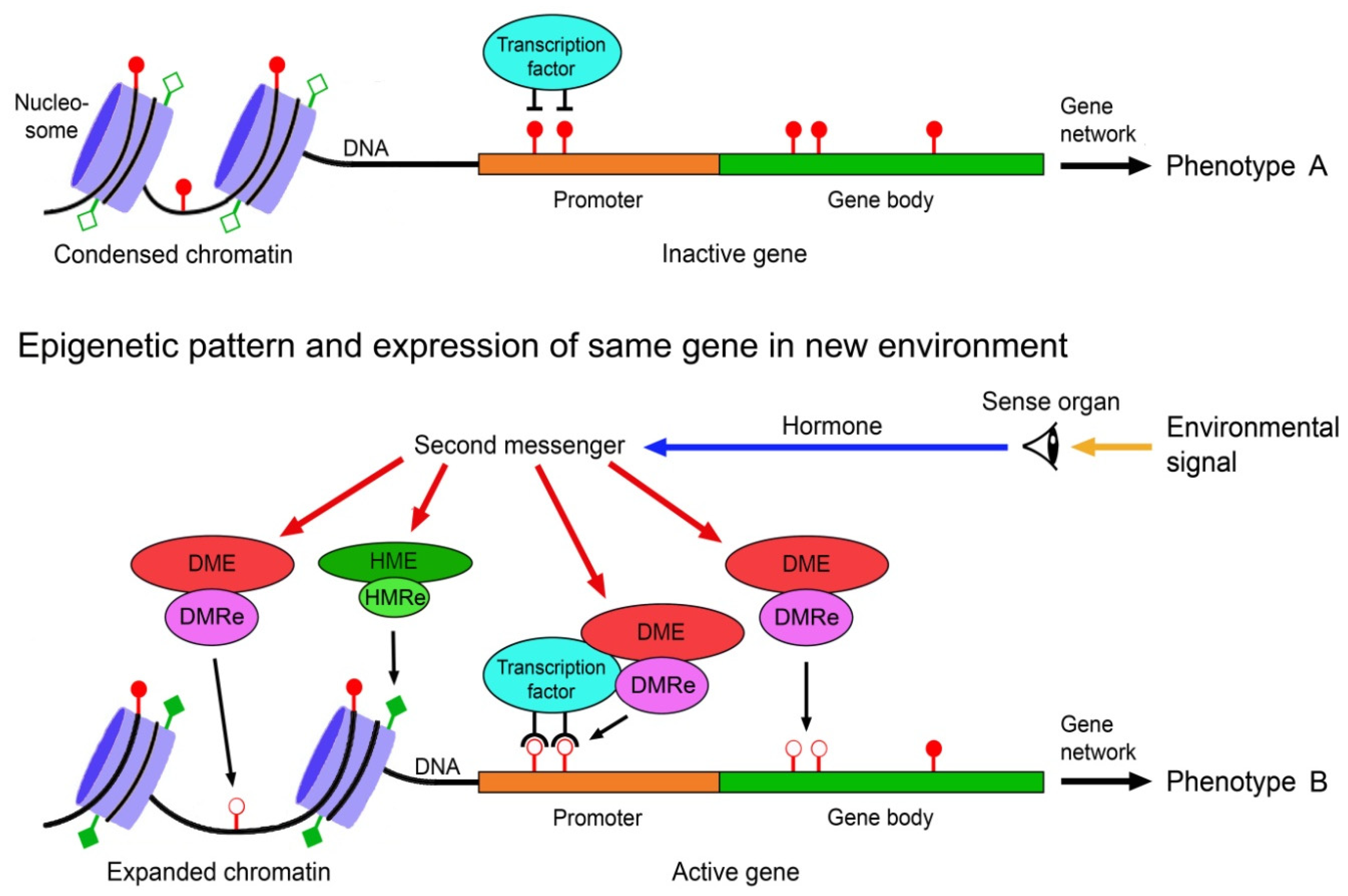
5. Relative Contributions of Genetic and Epigenetic Mechanisms to Phenotypic Diversity in Natural Populations
The relative contributions of genetic and epigenetic mechanisms to the production of phenotypic diversity in populations depends on several factors, including mode of reproduction, lifestyle and life history parameters. Since these factors vary significantly between and within animals, plants, fungi, protists and bacteria, different higher taxa and species may profit differently from genetic and epigenetic mechanisms. In asexually reproducing species, phenotypic variation is almost exclusively based on epigenetic mechanisms, whereas in sexual reproducers it relies on both genetic and epigenetic sources. In the latter, genetic mechanismsvariation may often be predominant.
The phenotypic diversity that can be generated by genetic mechanisms is also much dependent on population size and generation time,. because evolutionarily relevant mutations occur during cell division in unicellular organisms and meiosis in multicellular organisms. Bacteria often Bacteria usually have extremely high population densities, e.g. 109 cells per mL in culture, and very short generation times of 20 min to a few hours. Thus, they can rapidly produce high degrees of phenotypic diversity by genetic mutations. In contrast, plants and animals have much lower population densities and generation times of 1 to 40 years and a few days to 15 years, respectively, enabling only relatively slow generation of phenotypic diversity by mutation and recombination.
PlTants are sessile and animals are mostly vagile with the exception of the Porifera, Cnidaria, Bryozoa, Bivalvia, Cirripedia, Tunicata and Pelmatozoa. Unlike vagile organisms, sessile species cannot evade unfavourable environmental conditions by migration. Therefore, in order to compensate environmental stress they may use epiking these considerations together, genetic mechanisms particularly intensely.
Takmay contring all buthese considerations together, genetic mechanisms are thought to contributee a relatively larger proportion to phenotypic diversity relatively more iin bacteria and protists than in plants and animals. Epigenetic mechanisms are effective in all organisms but armay be particularly relevant for asexually reproducing, plants and animals and sessile and long-lived plants and animalstaxa that cannot evade unfavourable conditions by migration.
References
- Rengefors, K.; Kremp, A.; Reusch, T.B.H.; Wood, A.W. Genetic diversity and evolution in eukaryotic phytoplankton: Revelations from population genetic studies. J. Plankton Res. 2017, 39, 165–179.
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.V.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105.
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Recombination: The good, the bad and the variable. Philos. Trans. R. Soc. Lond. B 2017, 372, 20170279.
- Low, K.B.; Porter, D.D. Modes of gene transfer and recombination in bacteria. Annu. Rev. Genet. 1978, 12, 249–287.
- Seymour, M.; Räsänen, K.; Kristjánsson, B.K. Drift versus selection as drivers of phenotypic divergence at small spatial scales: The case of Belgjarskógur threespine stickleback. Ecol. Evol. 2019, 9, 8133–8145.
- Clegg, S.M.; Phillimore, A.B. The influence of gene flow and drift on genetic and phenotypic divergence in two species of Zosterops in Vanuatu. Philos. Trans. R. Soc. Lond. B 2010, 365, 1077–1092.
- Verhoeven, K.J.F.; Preite, V. Epigenetic variation in asexually reproducing organisms. Evolution 2014, 68, 644–655.
- Vogt, G. Epigenetic variation in animal populations: Sources, extent, phenotypic implications, and ecological and evolutionary relevance. J. Biosci. 2021, 46, 24.
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Frias-Laserre, D.; Villagra, C.A. The importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Front. Microbiol. 2017, 8, 2483.
- Vogt, G. Evolution, Functions and Dynamics of Epigenetic Mechanisms in Animals. In Handbook of Epigenetics: The New Molecular and Medical Genetics, 3rd ed.; Tollefsbol, T., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 521–549.
- Maeji, H.; Nishimura, T. Epigenetic mechanisms in plants. Adv. Bot. Res. 2018, 88, 21–47.
- Madhani, H.D. Unbelievable but true: Epigenetics and chromatin in fungi. Trends Genet. 2021, 37, 12–20.
- Sánchez-Romero, M.A.; Casadesús, J. The bacterial epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20.
- Gentilini, D.; Garagnani, P.; Pisoni, S.; Bacalini, M.G.; Calzari, L.; Mari, D.; Vitale, G.; Franceschi, C.; Di Blasio, A.M. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging 2015, 7, 568–576.
- Plotnikova, O.; Baranova, A.; Skoblov, M. Comprehensive analysis of human microRNA–mRNA interactome. Front. Genet. 2019, 10, 933.
- Nasrullah, A.H.; Ahmed, S.; Rasool, M.; Shah, A.J. DNA methylation across the tree of life, from micro to macro-organism. Bioengineered 2022, 13, 1666–1685.
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492.
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326.
- Neri, F.; Rapelli, S.; Kreplova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77.
- Gatzmann, F.; Falckenhayn, C.; Gutekunst, J.; Hanna, K.; Raddatz, G.; Coutinho Carneiro, V.; Lyko, F. The methylome of the marbled crayfish links gene body methylation to stable expression of poorly accessible genes. Epigenetics Chromatin 2018, 11, 57.
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92.
- Wu, H.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534.
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44.
- He, C.; Zhang, Z.; Li, B.; Tian, S. The pattern and function of DNA methylation in fungal plant pathogens. Microorganisms 2020, 8, 227.
- Nai, Y.-S.; Huang, Y.-C.; Yen, M.-R.; Chen, P.-Y. Diversity of fungal DNA methyltransferases and their association with DNA methylation patterns. Front. Microbiol. 2021, 11, 616922.
- Sánchez-Romero, M.A.; Cota, I.; Casadesús, J. DNA methylation in bacteria: From the methyl group to the methylome. Curr. Opin. Microbiol. 2015, 25, 9–16.
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500.
- Marmorstein, R.; Zhou, M.-M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2015, 6, a018762.
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281.
- Zhao, T.; Zhan, Z.; Jiang, D. Histone modifications and their regulatory roles in plant development and environmental memory. J. Genet. Genomics 2019, 46, 467–476.
- Brosch, G.; Loidl, P.; Graessle, S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol. Rev. 2008, 32, 409–439.
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110.
- Moutinho, C.; Esteller, M. MicroRNAs and epigenetics. Adv. Cancer Res. 2017, 135, 189–220.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 40.
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84.
- Senti, K.A.; Brennecke, J. The piRNA pathway: A fly’s perspective on the guardian of the genome. Trends Genet. 2010, 26, 499–509.
- Li, M.-Z.; Xiao, H.-M.; He, K.; Li, F. Progress and prospects of noncoding RNAs in insects. J. Integr. Agric. 2019, 18, 729–747.
- Wang, Y.; Xu, T.; He, W.; Shen, M.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics 2018, 110, 35–42.
- Waititu, J.K.; Zhang, C.; Liu, J.; Wang, H. Plant non-coding RNAs: Origin, biogenesis, mode of action and their roles in abiotic stress. Int. J. Mol. Sci. 2020, 21, 8401.
- Dhingra, S. Role of non-coding RNAs in fungal pathogenesis and antifungal drug responses. Curr. Clin. Microbiol. Rep. 2020, 7, 133–141.
- Stav, S.; Atilho, R.M.; Mirihana Arachchilage, G.; Nguyen, G.; Higgs, G.; Breaker, R.R. Genome-wide discovery of structured noncoding RNAs in bacteria. BMC Microbiol. 2019, 19, 66.
- Steffen, P.A.; Ringrose, L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 2014, 15, 340–356.
- Ciabrelli, F.; Comoglio, F.; Fellous, S.; Bonev, B.; Ninova, M.; Szabo, Q.; Xuéreb, A.; Klopp, C.; Aravin, A.; Paro, R.; et al. Stable Polycomb-dependent transgenerational inheritance of chromatin states in Drosophila. Nat. Genet. 2017, 49, 876–886.
- Wright, C.J.; Smith, C.W.J.; Jiggins, C.D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022, 23, 697–710.
- Zhang, J.; Zhang, Y.-Z.; Jiang, J.; Duan, C.-G. The crosstalk between epigenetic mechanisms and alternative RNA processing regulation. Front. Genet. 2020, 11, 998.
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing – immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490.
- Zhao, L.-Y.; Song, J.; Liu, Y.; Song, C.-X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808.
- Feinberg, A.P.; Irizarry, R.A. Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. S1), 1757–1764.
- Angers, B.; Perez, M.; Menicucci, T.; Leung, C. Sources of epigenetic variation and their applications in natural populations. Evol. Appl. 2020, 13, 1262–1278.
- Vogt, G. Disentangling the Environmentally Induced and Stochastic Developmental Components of Phenotypic Variation. In Phenotypic Switching: Implications in Biology and Medicine; Levine, H., Jolly, M.K., Kulkarni, P., Nanjundiah, V., Eds.; Academic Press: San Diego, CA, USA, 2020; pp. 207–251.
- Shah, J.M. Epimutations and mutations, nurturing phenotypic diversity. Genetica 2022, 150, 171–181.
- Vogt, G. Facilitation of environmental adaptation and evolution by epigenetic phenotype variation: Insights from clonal, invasive, polyploid, and domesticated animals. Environ. Epigenet. 2017, 3, dvx002.
- Leung, C.; Breton, S.; Angers, B. Facing environmental predictability with different sources of epigenetic variation. Ecol. Evol. 2016, 6, 5234–5245.
- Van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681.
- Skinner, M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinol. 2014, 398, 4–12.
- Xue, Y.; Acar, M. Mechanisms for the epigenetic inheritance of stress response in single cells. Curr. Genet. 2018, 64, 1221–1228.
- Liu, J.; He, Z. Small DNA methylation, big player in plant abiotic stress responses and memory. Front. Plant Sci. 2020, 11, 595603.
- Vogt, G. Epigenetics and Phenotypic Plasticity in Animals. In Epigenetics, Development, Ecology and Evolution; Vaschetto, L.M., Ed.; Springer: Cham, Switzerland, 2022; pp. 35–108.
- Foquet, B.; Castellanos, A.A.; Song, H. Comparative analysis of phenotypic plasticity sheds light on the evolution and molecular underpinnings of locust phase polyphenism. Sci. Rep. 2021, 11, 11925.
- Zhu, T.; Brown, A.P.; Ji, H. The emerging role of ten-eleven translocation 1 in epigenetic responses to environmental exposures. Epigenetics Insights 2020, 13, 1–9.
- Voigt, S.; Kost, L. Differences in temperature-sensitive expression of PcG regulated genes among natural populations of Drosophila melanogaster. G3 Gene Genome Genet. 2021, 11, jkab237.
- Danisman, S. TCP transcription factors at the interface between environmental challenges and the plant’s growth responses. Front. Plant Sci. 2016, 7, 1930.
- Huang, Q.; Ma, C.; Chen, L.; Luo, D.; Chen, R.; Liang, F. Mechanistic insights into the interaction between transcription factors and epigenetic modifications and the contribution to the development of obesity. Front. Endocrinol. 2018, 9, 370.
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018, 16, 1419.
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073.
- Kribelbauer, J.F.; Lu, C.-J.; Rohs, R.; Mann, R.S.; Bussemaker, H.J. Toward a mechanistic understanding of DNA methylation readout by transcription factors. J. Mol. Biol. 2020, 432, 1801–1815.