Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Neda Ali Al Bati and Version 2 by Sirius Huang.
Numerous efforts have been made to comprehend the pathogenic and host variables that contribute to COVID-19 susceptibility and pathogenesis. One of these endeavours is understanding the host genetic factors predisposing an individual to COVID-19. Genome-Wide Association Studies (GWAS) have demonstrated the host predisposition factors in different populations. These factors are involved in the appropriate immune response, their imbalance influences susceptibility or resistance to viral infection.
- coronaviruses
- COVID-19
- Genetic resistance
- protective immunity
1. Introduction
Specific Inborn Errors of Immunity (IEIs) have determined vulnerability towards acute COVID-19 since the outbreak of the COVID-19 pandemic. The COVID-19 Human Genomic Effort found IEIs to span eight genomic loci controlling the induction of type I Interferon (IFN) via TLR3 and IRF7 in 23 critically sick individuals [1][6]. Four unrelated and formerly healthy individuals observed autosomal and recessive mutations in the genetic loci encoding IRF7 or IFNAR1, leading to its deficiency. People with IEIs reveal that a type I IFN immune response is required to control COVID-19 infection. This observation led to the finding of already existing neutralising autoantibodies to type I IFNs, which denote IEIs related to type I IFN [2][1]. According to subsequent research in independent cohorts, over 10% of people with severe COVID-19 had neutralising autoantibodies against type I IFNs. According to a consortium, autoantibodies balancing the physiological amounts of type I IFNs are present in around 20% of individuals above 70 years-old, and with severe pneumonia [3][7]. Furthermore, a research consortium found that roughly 1% of male patients with severe pneumonia under 60 exhibited X-recessive TLR7 deficiency [4][8]. Before exposure to SARS-CoV-2, the people with IEIs and those with autoantibodies had no particular sensitivity to other severe viral illnesses.
This observation is in line with the fact that SARS-CoV-2 produces fewer type I IFNs than, say, the annual influenza virus [5][9]. However, one third of the adverse responses to the live attenuated yellow fever virus vaccination have been linked to type I IFN autoantibodies [6][10]. These cases show how the genetic explanation of an immune impairment in a few unusual individuals might reveal a system that is impaired in several people due to other factors. The number of people who display natural resistance to SARS-CoV-2 infection is unclear. Still, several lines of evidence have pointed to several candidate genes that may be implicated in the innate immune response that leads to the resistance towards SARS-CoV-2 infection.
Genome-wide association studies (GWAS) were used to find the ABO gene [7][8][11,12]. Although early findings related to the influence of blood group on COVID-19 intensity displayed a diverse form, a recent meta-analysis involving approximately 50,000 participants from 46 studies indicated that this locus affected infection vulnerability. On the other hand, the O allele has a minor protective effect, with an odds ratio of only ~0.90. ABO blood types may also function as co-receptors for SARS-CoV-2, even though no unifying resistance mechanism has yet been proposed [7][9][11,13]. Pernio (chilblain) linked with the SARS-CoV-2 pandemic is an uncommon symptom in those who have been exposed to the virus, but it may provide knowledge about infection resistance mechanisms [10][11][14,15]. The skin lesions observed in familial chilblain lupus and Aicardi–Goutières syndrome, which are monogenic illnesses characterised by gene variations leading to an enhanced type I IFN response, are mimicked by pandemic-associated pernio (‘COVID toes’) [12][16]. Although most people with pernio are seronegative, skin biopsy specimens have shown the presence of the spike protein of SARS-CoV-2 and a robust local type I IFN response, suggesting early viral clearance [13][17]. These findings point to the existence of infection and, as a result, the lack of natural infection resistance. However, through a better understanding of this process, researchers may obtain an insight into the host mechanisms that limit viral replication and promote resistance to COVID-19 infection.
Additional putative host genes that support the life cycle of the COVID-19 virus have been found by in vitro interactome analyses. COVID-19 infection requires the presence of an ACE2 receptor for entry into the host cell and the serine protease, TMPRSS2, for the priming of spike protein, which was found early in the pandemic [14][15][16][17][18,19,20,21]. Likewise, GWAS discovered an uncommon variation near ACE2 that is found to protect against SARS-CoV-2 infection, probably by lowering ACE2 expression [18][22]. Additionally, specific human ACE2 allelic variants bind the spike protein of SARS-CoV-2 with varying affinities, although their influence on infection is unclear [19][23]. In addition, a genome-wide CRISPR knockout screen identified TMEM41B as essential for SARS-CoV-2 and other coronaviruses infection [20][24]. Flaviviruses also need the endoplasmic reticulum transmembrane protein TMEM41B. Its influence on COVID-19 infection is unclear. However, an allele detected in East and South Asians was shown to cause reduced ability to sustain flavivirus infection [21][25].
Similarly, a combined affinity purification approach and mass spectrometry of human proteins associated with SARS-CoV-2 led to a massive protein-protein interaction map [22][26]. As a result of the functional study of this interactome, a list of important host variables for the transmission of COVID-19 was created [23][27]. Even though there isn’t any human research that links this interactome to the risk of infection, the genes involved and the loci found by GWAS can be looked at as possible places to find genetic variants that protect against infection.
2. Genetic Resistance to Virus Entry
Individuals with allelic variants of CCR5 that result in CCR5 deletion, termed “elite resistors” to the human immunodeficiency virus (HIV), are a suitable example of genetic resistance due to a lack of viral receptors on host cells. Interestingly, there is a predominance of this CCR5 mutation in Europeans. This might be influenced by the fact that it confers a reduced smallpox death rate [24][25][26][27][51,52,53,54]. It is unknown whether deletion of CCR5 precludes co-reception of the variola virus or lowers the viral-induced lethal immune response by inhibiting inflammatory chemokine signals. Moreover, acquiring mutations in the spike protein allowed it to bind with greater affinity to the human host cell receptors, ACE2 for SARS-CoV-2 and SARS-CoV or DPP4 for the Middle East respiratory syndrome-related coronavirus (MERS-CoV). This was a crucial step in the transition of coronaviruses from bats to humans [28][2].
As per previous studies, plant breeders recognised that genetic resistance to communicable illness is unstable over time; therefore, variant bacteria can be selected rapidly, escape resistance and spread better, whether through loss of receptors or by developing immunological responses [29][30][55,56]. Every year, the insecure resistance state is focused on preventing viral transmission. This is why we require a new seasonal influenza vaccination each year. Influenza elicits long-lasting neutralising antibodies, resulting in a stronger selection for the antigenic drift in virus epitopes, causing them to escape being recognised by the current set of antibodies. Coronaviruses possess the largest genomes of any RNA viruses, and, contrary to other RNA viruses, they copy genomes with a considerably greater degree of accuracy [31][57]. The COVID-19 virus develops point mutations at a slower rate of one in ten thousand bases yearly [32][58]. As a result, coronaviruses do not use antigenic drift as a technique to avoid neutralising antibody development, unlike influenza and human immunodeficiency virus vaccines. Instead, after human infection with the common cold coronavirus HCoV-229E [33][34][59,60] or severe conditions with SARS-CoV, neutralising antibody production is low and oddly brief [35][36][61,62]. In the poultry sector, attenuated vaccines against contagious coronavirus use are limited due to short-lived antibody production [37][38][63,64]. On the one hand, influenza evades the neutralising antibody yield due to a higher rate of mutations in the viral genome, while coronavirus alters the body’s mechanisms for neutralising antibody responses, leading to short-lived coronaviruses that do not halt viral spread in the population [34][60]. Figure 1 illustrates the mechanism of genetic resistance of the host against virus entry.
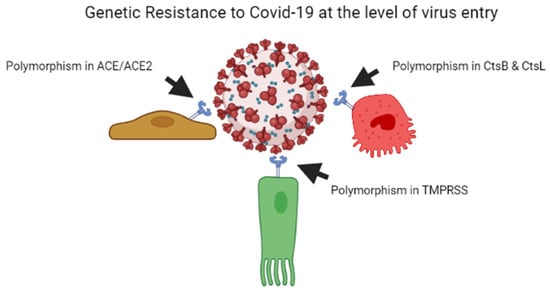
Figure 1.
Genetic resistance of host against the COVID-19 virus at the level of entry of the virus.
2.1. Angiotensin Converting Enzymes
Angiotensin I-converting enzyme (ACE) and angiotensin-converting enzyme-2 (ACE2) represent a pair of homologous genes that govern the physiological balance of the renin-angiotensin system (RAS). SARS-CoV-2 uses ACE2 receptors to infect susceptible cells [17][21]. In numerous methods, ACE and ACE2 influence each other’s expression levels [39][65]. Therefore, examining the genetic variations of these genes can assist us to determine why COVID-19 is more acute in certain individuals.
2.1.1. Angiotensin-I Converting Enzyme
The insertion or deletion of a 287 bp Alu repeat is a frequent polymorphism of the ACE gene, and the DD allele is linked to increased ACE levels [40][66]. The relevance of the ACE/ACE2 balance in COVID-19 development and treatment has received much attention [40][41][42][43][66,67,68,69]. ACE and ACE2 balance the local vasodilator/antiproliferative and vasoconstrictor/proliferative activities of the RAS system [40][66]. Tissue damage, fibrosis, thrombosis, proliferation and inflammation may all be exacerbated if the ACE/ACE2 balance is disrupted. Thus, in contrast to ACE2, the ACE gene sequence may influence the results of the COVID-19 clinical trial [44][45][70,71]. Delanghe et al. found that the incidence of COVID-19 infections was adversely correlated with the frequency of the D allele in 25 European countries. Similar results were observed in 33 European, North African, and Middle Eastern nations, indicated that increasing D allele frequency was related with a decline in COVID-19 incidence but an increase in mortality [46][72]. In European and Asian nations, however, greater frequency of the I/I genotype was negatively associated with vulnerability to SARS-CoV-2 infection and subsequent death [47][73].
Similarly, an ecological analysis found that having a higher I/I genotype frequency was linked to lower COVID-19 mortality in 25 nations worldwide [48][74]. In Asian populations, numbers of the D allele were determined by the number of SARS-CoV-2 infected patients per million [49][75]. In Asian populations, mortality rates and the presence of the D allele revealed the substantial positive connection, demonstrating that greater levels of ACE are harmful to COVID-19 patients [49][75]. COVID-19 infection and fatality rates are both greater in the European population [47][73]. Since, the ACE DD genotype is more common in Europeans than in Asians, higher death rates in Europeans are likely to be accounted by ethnic variations in ACE I/D polymorphism allele prevalence.
2.1.2. Angiotensin Converting Enzyme-2
Angiotensin-I and Angiotensin-II are cleaved into peptides such as Angiotensin 1–9 and Angiotensin 1–7, respectively, by ACE2, the master regulator of the RAS. These peptides are essential components in cardiovascular physiology control [50][51][76,77]. SARS-Cov-2 infection suppresses ACE2 expression and interferes with its homeostatic and defensive actions, causing inflammation [52][78]. ACE2 is present at various levels in most human tissues [53][54][79,80]. As SARS-CoV-2 attacks alveolar epithelial cells via the ACE2 receptor, the ACE2 expression levels in different organs might reveal genetic sensitivity to COVID-19 [55][56][81,82]. Single-cell RNA sequencing of various cell types in lung tissue revealed that bronchial branches contain a transient cell population with the ACE2 phenotype. Among these cells, 40% also express the transmembrane serine protease 2 (TMPRSS2) involved in the priming of the viral spike protein, thus making these cells more vulnerable to COVID-19 infection [57][83].
The ACE2 gene contains the regulatory elements for chromatin alteration and transcription factors that regulate expression levels epigenetically and hormonally. In several tissues, there is a link between ACE2 expression and age, gender, ethnicity, and BMI [58][84]. Asian females have the most significant connection with age, followed by sex and ethnic groupings. In a study of 305 people, the expression of the ACE2 gene in the nasal epithelium was explored, and it was discovered that younger children under the age of ten have the lowest ACE2 levels [59][85]. As a result, it was hypothesised that the reduced risk in children was linked to ACE2 expression levels that decreased with age [59][85]. Androgen receptor signaling governs the transcription of both ACE2 and TMPRSS2. Therefore, it is believed that androgen receptors regulate the production of ACE2 and TMPRSS2. This is the reason behind the gender differences in COVID-19 severity and the polymorphism in the androgen receptor linked to the condition [60][86]. ACE2 is substantially expressed in severe COVID-19 patients compared to controls, according to transcriptome analysis of 700 lung samples. More than one illness at the same time (co-morbidity) may increase the risk of serious COVID-19 [55][81]. On the other hand, other investigations have demonstrated a correlation between ACE2 expression and COVID-19 severity [58][61][62][84,87,88].
2.1.3. Genetic Variants of ACE2
The genotype of ACE2 is linked to the binding affinity and structure of the protein, as well as serum concentration and systemic angiotensin levels [63][89]. In the NCBI database, about 18,000 single nucleotide variations of human ACE2 are denoted. From genetic susceptibility to COVID-19 infection, a comparative genomic investigation of ACE in different populations was conducted. Cao and his co-workers evaluated about 1700 ACE2 gene variations from different databases and the distribution system of expression quantitative trait loci (eQTLs) from the genotype and expression data of different tissues [64][90]. The investigating groups found various allele frequencies of ACE2 coding across the different populations (South Asian, East Asian, African, European, and mixed American populations). The allele frequencies of 11 of the 15 eQTLs that were linked to ACE2 expression were greater in East Asians (0.73–0.99) than in Europeans (0.44–0.65), which is indicative of the differential susceptibility to SARS-CoV-2 among different cultures [64][90]. The frequency of uncommon variations in the host genes encoding for virus entry machinery (ACE2, CtsB, CtsL, and TMPRSS2) are very variable among groups, suggesting that they might be crucial in SARS-CoV-2 entrance [65][91]. It was discovered that 13 genetic variations in ACE can improve the interaction among ACE2 and the viral S1 protein. The Europeans and Africans varied considerably with respect to rs73635825 (S19P), rs1244687367 (I21T), rs4646116 (K26R), rs781255386 (T27A), rs1199100713 (N64K), and rs142984500 (H378R) variations [65][91]. Various databases, and around 81000 human genomes, were used to look for ACE2 and TMPRSS2 polymorphisms [66][92]. The distribution of harmful mutations in ACE2 varied significantly between nine groups. African/African American (AFR) and Non-Finnish European (EUR) populations, for example, had 39% and 54% deleterious variations, respectively. The p.Met383Thr and p.Asp427Tyr variations was found in AFR groups, whereas the p.Pro389 his variation was found in Latino/Admixed American communities and was characterised as an inhibitory variant towards the interaction with the SARS-CoV-1 spike protein [66][92]. Gibson and his coworkers identified uncommon variations in distinct groups likely to impact spike protein binding [67][93]. They discovered that some exceptional variants are more common in certain groups or genders. For instance, the rs4646116 (p.Lys26Arg) allele is found at a greater frequency in Ashkenazi Jewish males than in EUR males; this allele was observed at higher frequencies in females. The difference in binding energy of the 15 ACE2 missense variations to SARS-CoV-2 indicates that Glu37Lys boosted binding with maximum efficiency, while Asn720Asp reduced it significantly. The N720 variation found near the TMPRSS2 cleavage site, is one of the most common mutations in Europe. The N720D variation alters the flexibility and stability of ACE2, and generates a preferred location for TMPRSS2 binding and cleavage, according to computational structural biology and molecular modelling [68][94]. As a result of the increased interaction between ACE2 and TMPRSS2, N720D carriers have higher S protein binding and viral entry [68][94].
Seventeen natural ACE2 coding variations were detected at the critical S protein binding sites in the natural ACE2 coding variants [69][95]. While most variants had the same binding affinity, the intermolecular interactions of rs143936283(E329G) and rs73635825(S19P) alleles were noticeably different. As a result, it is believed that the rs143936283 and rs73635825 alleles conferred resistance to SARS-CoV-2 attachment to the human ACE2 receptor [69][95]. K26R and I468V variations can impact the binding properties of S proteins by enhancing binding free-energy and lowering the binding affinity, according to molecular dynamic simulations [54][80]. Non-Finnish Europeans are more likely to have the K26R variation mutated, whereas East Asians are more likely to have the I468V variant mutated [54][80]. Whole exome sequencing (WES) data from 6930 healthy Italian persons were used to identify potential variations that affect protein stability [70][96]. Missense variations p.(Asn720Asp), p.(Lys26Arg), and p.(Gly211Arg) were prevalent and anticipated to interfere with the structure and stability of ACE2 protein, whereas p.(Pro389His) and p.(Leu351Val) were found to be uncommon and predicted to interfere with the binding to the viral spike protein. Moreover, it was observed that the control group had statistically significant higher allelic variability, indicating that genetic background may be responsible for the individual variations related to COVID-19 [70][96].
A large genomic dataset determined nine ACE2 variations anticipated to enhance susceptibility, and 17 projected to demonstrate reduced binding towards S protein and protection against SARS-CoV-2 transmission [71][97].
In contrast to these findings, a few studies claimed no link between ACE2 polymorphisms and illness severity [72][98]. WES investigated ACE2 genetic variations in 131 DNA samples from COVID-19 patients from a hospital in Italy, compared to a control group of 1000 people [73][99]. There was substantial variation in the frequency of the c.1888G>C p.(Asp630His) mutation across ethnically matched groups; nonetheless, there was no link seen between ACE2 variants and COVID-19 severity [73][99]. In silico simulations of ACE2-S1 protein binding kinetics have also shown some discrepancies [54][65][66][68][69][70][80,91,92,94,95,96]. The relevance of ACE2 genotypes in COVID-19 might be better understood with further functional investigations and genotype analysis.
2.1.4. Dipeptidyl Peptidase
Dipeptidyl Peptidase, also known as DPP4, is a critical human protein involved in numerous peptide interactions. This protein has a significant function in regulating diabetes [74][101]. However, it also interacts with several viral proteins [74][101]. A recent study showed that mutations in DPP4 were more evident in COVID-19 asymptomatic patients [75][46]. The severity of COVID-19 was also correlated with the down-regulation of DPP4 [76][102]. DPP4 expression was also associated with obesity, and subsequent investigations validated its association with COVID-19 [77][78][79][80][103,104,105,106]. In one study, missense and splice acceptor variants in DPP4 (c.95-2A > G, c.796G > A, c.1887 + 3G > A) were reported in COVID-19 patients and related to the severity of the disease [81][107].
2.1.5. Furin
Cleavage is performed by furin, and a study determined that the cleavage site is critical for the entrance of the virus. The entry mechanism of SARS-CoV-2 into the host depends upon the cleavage of spike glycoprotein at a specific site [82][108]. Several mutations [11][15] were identified in the furin protein, and a few, such as R37C, R81C, R86Q, R637Q, R677W, R745Q, and S685P [3][7], showed a direct impact in lowering the risk of virus progression [83][109].