Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Enol Lopez.
Coumarin and its derivatives have significantly attracted the attention of medicinal chemists and chemical biologists due to their huge range of biological, and in particular, pharmacological properties. Interesting families of coumarins have been found from marine sources, which has accelerated the drug discovery process by inspiring innovation or even by the identification of analogues with remarkable biological properties.
- coumarin
- biological activity
- marine natural products
1. Simple Coumarins
Simple substituted coumarins are the structurally less complex class of coumarins. The scaffold is constituted by a bicyclic system and different substitution patterns at the C-3, C-4, C-5, C-6, C-7 and C-8 positions. Due to the already defined biological activity [28,29], two compounds—umbelliferone (7, R7 = OH) and scopoletin (8, R6 = OCH3 and R7 = OH) —are highlighted here (Figure 2). These molecules were isolated in 2012 from the leaves of the mangrove endophytic fungus Penicillium sp. ZH16 from the South China sea [30].
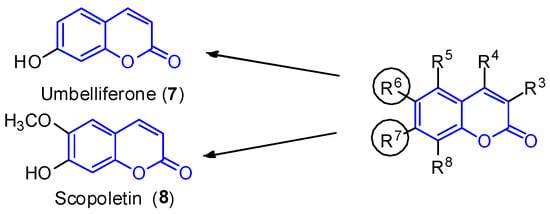
Figure 2.
Simple coumarin derivatives: umbelliferone (
7
) and scopoletin (
8
).
Umbelliferone (7) shows anti-inflammatory [31] and antitumoral activities [32]. In 2015, Yu et al. reported the anticancer activity of umbelliferone (7) against HepG2 cancer cells, inducing apoptosis in cells [33], whereas scopoletin (8) inhibits PC3 proliferation, a human prostate cancer cell line [34]. Additionally, both compounds exhibit anti-acetylcholinesterase (AChE) [35,36] and antioxidant activities [37].
As a result of the importance of these scaffolds in organic and medical chemistry, many synthetic routes to obtain simple substituted coumarins have been explored over the years [38,39]. The most classical strategies involve Knoevenagel [40], Pechmann [41] and Perkin [42] condensations, intramolecular [43,44] and intermolecular Wittig reactions [45], ring-closing metathesis [46], as well as different reactions between the corresponding salicylaldehydes with ketene [47] or arylacetonitriles (18) [48] (Scheme 1).
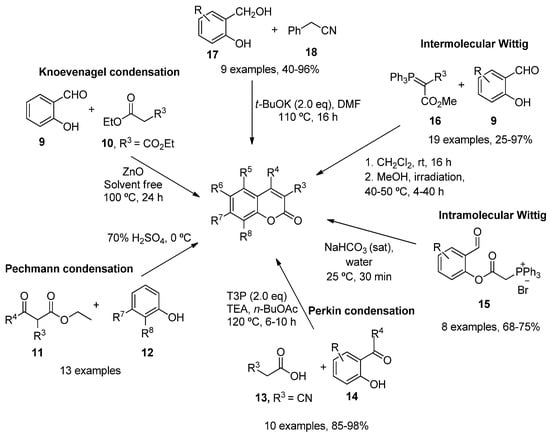
Scheme 1.
Synthetic approaches in the preparation of functionalized coumarins.
Over the last few years, transition metal catalysis, involving palladium [49,50], rhodium [51], iron [52] or cobalt [53], has also been used to synthesize different coumarin derivatives. In addition, modern methodologies such as microwave irradiation [54,55], flow chemistry [56], photochemistry [57], ionic liquids [58,59] and organocatalytic reactions [60], proved to be very effective.
For instance, Y. Li et al. employed the photocatalytic isomerization of ortho-E-hydroxycinnamates (19) to generate Z isomers, which underwent lactonization to provide coumarin compounds in high yields (Scheme 2) [61]. A similar strategy was developed by Shu et al. but using Ir2(ppy)4Cl2 as the catalyst (Scheme 2) [62].
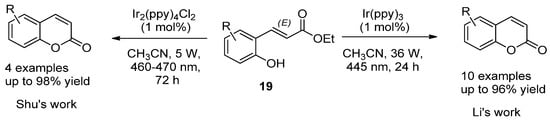
Scheme 2.
Photocatalytic reactions in the synthesis of coumarins.
Au(I)-catalysts have also been screened to synthesize coumarins by the intramolecular arylation (IMHA) of phenol-derived propiolates (20) [63]. IMHA reactions were carried out using Echavarren’s catalyst (21), (acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate, to give numerous derivatized compounds in high yields (Scheme 3).
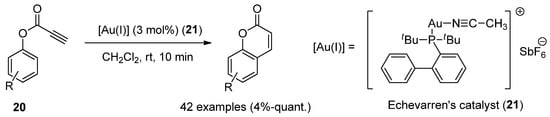
Scheme 3.
Gold(I)-catalyzed reaction in the preparation of coumarin derivatives.
Furthermore, other metal-free methodologies have been reported. In 2016, Lee et al. reported a TfOH-mediated condensation of phenols (22) with propiolic acids (23), followed by intramolecular arylation [64], which was applied to obtain natural umbelliferone (7) in an 81% yield (Scheme 4).
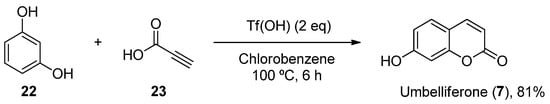
Scheme 4.
Metal-free methodology for the preparation of umbelliferone (
7
).
2. 3-Substituted (Imino- and Amido-) Coumarins
2.1. 3-Iminocoumarins
All 3-iminocoumarins (24–39) reported in the literature have been isolated from mangrove fungi present in the South China sea, along with tens of other metabolites [65]. To the best of our knowledge, these compounds do not show relevant biological activity, and no synthetic approaches have been published to date (Figure 3).
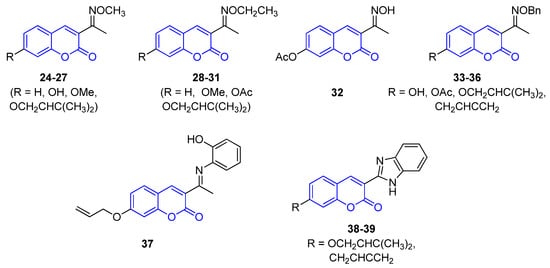
Figure 3.
Representative examples of iminocoumarins from marine sources.
2.2. 3-Amidocoumarins
Trichodermamide A (40), B (41) and aspergillazine A (42) were the first 3-amidocoumarins isolated at the beginning of the 21st century from different marine-derived fungi, Trichoderma virens and Spicaria elegans [66,67,68]. Spectroscopic analysis and chemical methods (a modified Mosher’s method) allowed for the determination of the structure and stereochemistry of 40 [66] and 42 [69], while the structure of 41 was established by X-ray diffraction analysis (Figure 4) [66].
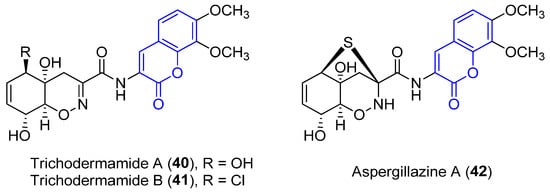
Figure 4.
Representative examples of amidocoumarins from marine sources.
Compounds 40 and 42 proved to display a weak cytotoxic activity against an HL-60 cell line (IC50 = 89 and 84 Μm, respectively) [67]. By contrast, as a result of the presence of the chlorohydrin group in 41, it displays in vitro cytotoxicity against HCT-116 colorectal cancer cells (IC50 = 0.32 μg/mL) [66] and HeLa cells (IC50 = 3.1 μM), by breaking double-stranded DNA [70]. Moreover, weak antimicrobial activities have been reported [68].
Recently, four new 3-amido compounds have been isolated (Figure 5). On the one hand, long-term static fermentation of the strain of Trichoderma sp. TPU199 (cf. Trichoderma brevicompactum) induced the production of dithioaspergillazine A (43), which possesses a disulphide bridge by comparison with spectroscopic data. In contrast to aspergillazine A (42), the compound inhibits the proliferation of the colon cancer HCT-15 cell line (IC50 = 13 μM) and Jurkat leukemia cells (IC50 = 1.3 μM) [71]. On the other hand, trichodermamide C (44) and hatsumamide A (45) and B (46) were isolated from the deep sea-derived fungus Penicillium steckii FKJ-0213 by physicochemical (PC) screening [72]. The structure of 44, which also contains a 1,2-oxazine system, was previously established by NMR, UV, IR, MS and X-ray diffraction data. It shows moderate cytotoxic effects towards human colorectal carcinoma HCT116 (IC50 = 0.68 μg/mL) and human lung carcinoma A549 (IC50 = 4.28 μg/mL) [73]. The structure and stereochemistry of 45 and 46 were elucidated by mass spectrometry, 1D and 2D NMR data (COSY, HMQC, HMBC and ROESY) and by comparing data with other already known compounds. No biological activity of 46 has been reported. However, 45 presents antimalarial activity against the K1 and FCR3 strains of Plasmodium falciparum, with IC50 values of 27.2 an 27.9 μM, respectively, and cytotoxicity against five human tumor cell lines, HeLa S3 (IC50 = 15.0 μM), HT29 (IC50 = 6.8 μM), A549 (IC50 = 13.7 μM), H1299 (IC50 = 18.7 μM) and Panc1 (IC50 = 12.9 μM) [72].
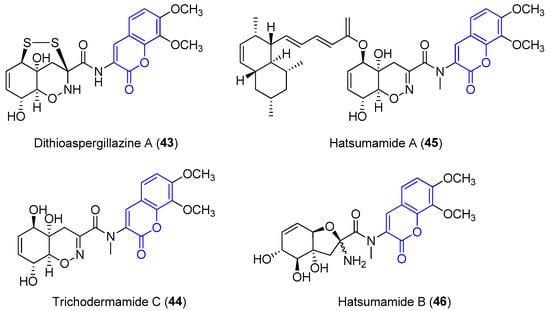
Figure 5.
Other examples of amidocoumarins from marine sources.
Trichodermamides 40, 41 and 44 could be disconnected into two fragments: an oxazine ring moiety 47 and an aminocoumarin 48 (Scheme 5). Different synthetic strategies have been developed in order to afford the 4H-5,6-dihydro-1,2-oxazine fragment 47.
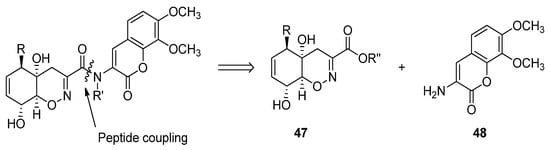
Scheme 5.
Retrosynthetic analysis for the preparation of trichodermamides.
In 2008, Joullié and Wan described the total synthesis of 40 and 41 [74]. Thus, the advanced intermediate 52, obtained in 18 linear steps from (–)-quinic acid 49, was treated with hydroxylamine to obtain an oxime, which in situ underwent an intramolecular epoxide ring opening upon addition of NaOH. Oxazine 53, obtained as a single diastereomer, was then converted (over four reaction steps) into enone 54. A Luche reduction followed by selective primary alcohol oxidation provided acid 55 in a good yield (Scheme 6).
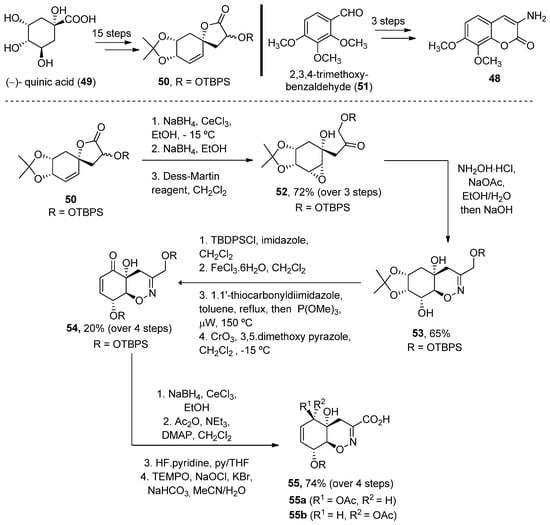
Scheme 6.
Methodology for the preparation of fragment
G
.
The coupling reaction between carboxylic acid 55 and aminocoumarin B (obtained in three steps from 3,4-trimethoxy-benzaldehyde) was performed using EDCI in 30% pyridine/dichloromethane. Compound 40 was obtained after coupling and two deprotection steps in a 53% yield, while 41 required an additional treatment with mesyl chloride in order to obtain the corresponding allylic chloride (32% over four reaction steps). The total enantioselective syntheses were achieved in 31/32 reaction steps, with an average of an 85% yield for each reaction step (Scheme 7) [74].
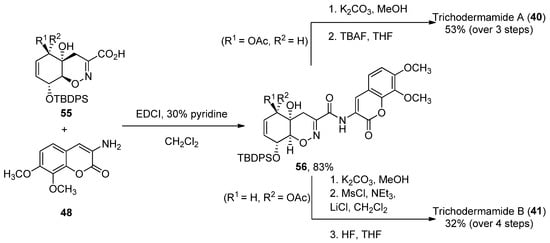
Scheme 7.
Final steps of the synthesis of trichodermamides A (
40
) and B (
41
).
More recently, a new concise total synthesis of trichodermamides A, B and C has been described [75]. A 1,2-addition of an αC-lithiated O-silyl ethyl pyruvate oxime 57 to benzoquine 58, followed by an oxa-Michael ring closure was applied to accomplish the formation of the cis-fused 1,2-oxazadecaline core 59 in a 92% yield. A modified Luche reduction and treatment with Pd(PPh3)4 in the presence of N,O-bis-(trimethylsilyl)acetamide (BTSA) was carried out to give dienol 60 in a high yield, a common intermediate in the synthesis of the three natural products (Scheme 8).
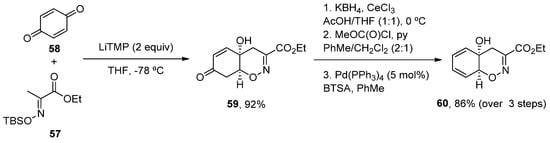
Scheme 8.
Methodology for the preparation of fragment
60
.
Once the oxazadecaline 60 was obtained, similar chemical steps, but arranged in different order, provided the three natural compounds. In these syntheses, the two key steps are the amide coupling, mediated by HATU in the presence of sym-collidine, and a final selenoxide [2,3]-sigmatropic rearrangement with H2O2 in pyridine [76], which was previously used by Zakarian and Lu [77]. Trichodermamides 40, 41 and 44 have been obtained after 9, 12 or 13 steps, respectively, in moderate yields (Scheme 9).
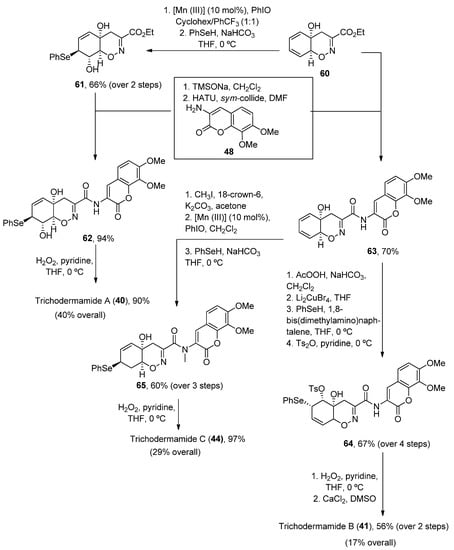
Scheme 9.
Synthetic methodology for the preparation of trichodermamides A (
40
), B (
41
) and C (
44
).
To the best of our knowledge, no synthetic approach for 42, 43, 45 and 46 has been reported yet.
3. Tricyclic Coumarins
3.1. Furocoumarins
First, furocoumarins (also called psoralenes) are described as coumarin derivatives with a fused furan ring with important biological activities, such as photoreactivity with DNA [78]. Four structures of furo[g]coumarins (66–69) were found in the endophytic fungus Penicillium sp. ZH16 from the South China sea (Figure 6) [30].
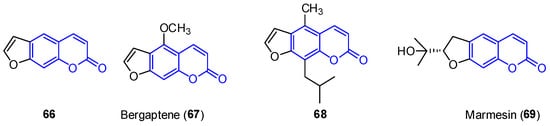
Figure 6.
Examples of furocoumarins from marine sources.
From this series, the derivative 68 was tested against KB and KBV200 cells demonstrating relevant cytotoxicity (IC50 of 5 and 10 μg/mL, respectively). In addition, 69 shows interesting photochemotherapeutic effects under near UV and blue light photosensitization (LD50 of 2 nM and 12 nM, respectively). These cytotoxic studies suggest the possibility of furocoumarins being involved in the high incidence of cancer in Nigeria [79].
The synthesis of furocoumarins has been known for a long time. However, novel methodologies have been developed during the last decades for the effective preparation of these compounds, many of which involve metal-catalyzed transformations that provide new structural diversity [80,81].
One such example has been the preparation of bergaptene (67). Although it was first isolated in 1834, it was not until 1936 when the first synthetic approach was described. Then, other methodologies were developed during the next decades [82]. Recently, Zhimin et al. reported a new synthesis of 67 in a higher isolated yield (55%) compared to the reported methodologies. Following known methodologies, phloroglucinol was used as the starting material to construct benzofuran-3-one 72 (by monomethylation and Pechmann reactions). Various conventional steps (acetylation, deacetylation) provided the intermediate 75 from which a fused lactone ring was constructed by acetylation and Pechmann condensation. A final dehydrogenation step with DDQ provided the final product 67 (Scheme 10).
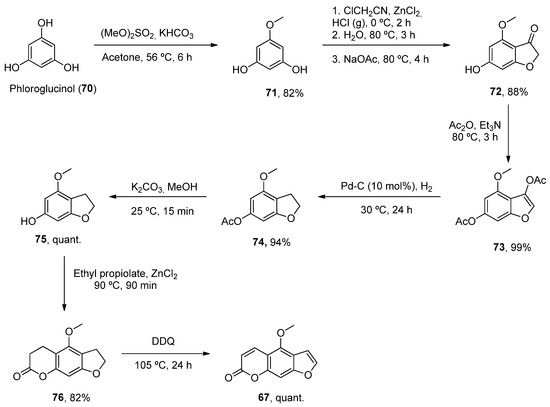
Scheme 10.
Synthetic methodology for the preparation of bergaptene (
67
).
3.2. Benzo[c]coumarins
Benzocoumarins are π-extended structures in which the coumarin core is fused with a benzene ring at different positions. Four benzo[c]coumarins (77–80, 3,4-benzocoumarins or alternariol derivatives) have been found from ocean sources, produced by the mangrove endophytic fungus No. 2240, from the South China sea coast (Figure 7) [83]. The structures were determined by spectroscopic analysis using NMR, IR and UV experiments and by comparison with the literature data.
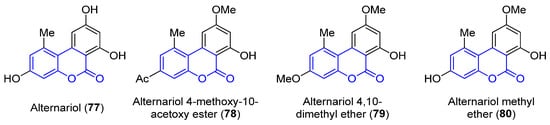
Figure 7.
Examples of benzo[c]coumarins.
Alternariol (77, R1 = R2 = OH) and its derivatives (78–80) have demonstrated mutagenic properties against the human epidermoic carcinoma KB and KBv200 cell lines. In particular, 77 and 80 show stronger IC50 values (3.17, 3.12 and 4.82, 4.94 μg/mL, respectively) in both cell lines, in comparison with the weaker activities found for the other compounds (IC50 > 50 μg/mL).
The construction of benzocoumarins depends on the location of hydroxyl and formyl groups on the starting material, which is normally hydroxynaphthaldehyde. Many complementary strategies have been reported for the general synthesis of benzo[c]coumarin derivatives, which are based on carbon–carbon and carbon–oxygen bond formation strategies or cyclization reactions [84].
A total synthesis of 77 was described independently by the Podlech and Kim groups [85,86]. The key step in both protocols was a Suzuki–Miyaura cross-coupling of boronic acid 81 with a brominated aldehyde 82 to obtain an advanced precursor 83. A final cyclization step was needed to obtain the final product 77 (Scheme 11).
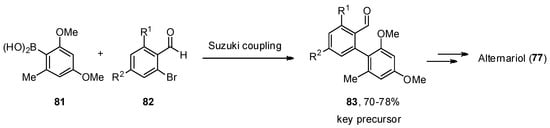
Scheme 11.
Critical key step in the total synthesis of alternariol (
77
).
More recently, the most common techniques in the synthesis of general benzo[c]coumarins have been the oxidative cyclization of biphenyl-2-carboxylic acid compounds [87] and Hurtley condensation [88]. However, other strategies that generate chemical diversity have been applied to obtain highly functionalized benzco[c]coumarins. For instance, Bodwell et al. prepared a set of benzo[c]coumarins by an inverse electron demand Diels–Alder reaction [89]. Later on, a multicomponent version (9, 84 and 85) comprising 6 reaction steps and increasing chemical diversity was disclosed by the same groups (Scheme 12) [90].
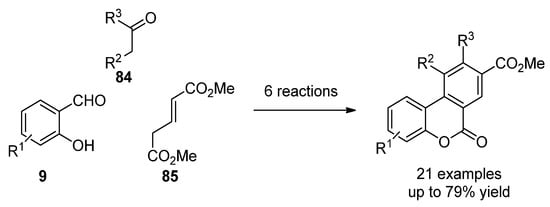
Scheme 12.
Multistep synthesis of benzo[c]coumarins.
Later, Lee’s group reported the reaction of hydroxychalcones (86) and β-ketoesters (87), in the presence of a base, in sequential Michael addition/intramolecular aldol condensation/oxidative aromatization/lactonization processes (Scheme 13) [91].
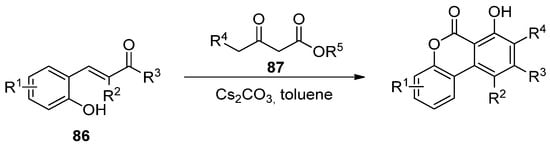
Scheme 13.
Domino reaction process for the synthesis of benzo[c]coumarins.
3.3. Other Tricyclic Coumarins
In addition to the previously mentioned groups, other tricyclic coumarins have been found in marine organisms. The limited number of their structural features does not allow for their classification in a particular group. Two pyrano[g]coumarins were isolated from Streptomyces violans bacteria and Ascomycete Leptosphaeria oraemaris fungi (compounds 87 and 88, respectively). In addition, iotrochotazine A (89) was found in the marine sponge Iotrochota sp. in Australia, and it is used as chemical probe to study Parkinson’s disease [92]. A series of dihydrocoumarins (90–95) was also found in Rhizophora stylosa mangrove trees in Okinawa, Japan. These compounds present DPPH free radical scavenging properties (EC50 4.6–10.3 μM) and serve as traditional medicine for the local people due to their antioxidant activities (Figure 8) [93,94].
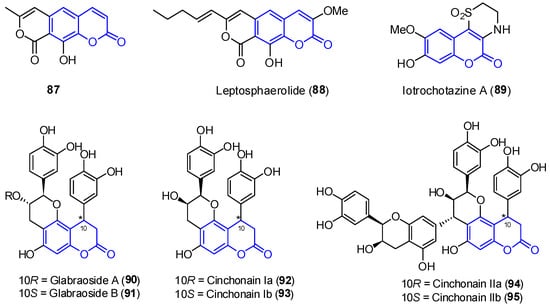
Figure 8.
Examples of tricyclic coumarins found from marine sources.
To the best of our knowledge, there are not many protocols reported for the preparation of each of these scaffolds. For instance, cinchonain derivatives 92 and 93 were successfully synthetized by the Kadota group, in a one-pot regioselective procedure involving a dienone–phenol rearrangement followed by a Michael-type reaction (Scheme 14) [95].
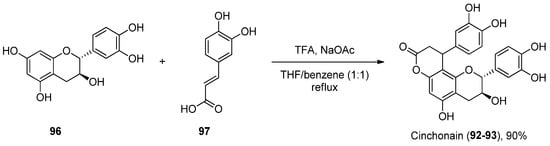
Scheme 14.
Synthesis of cinchonain.
Considering 89, a total synthesis was developed in 2014 through a one-pot enamine formation/intramolecular conjugate addition/oxidation sequence. The confirmation of the natural product allowed for subsequent biological investigations (Scheme 15) [92].
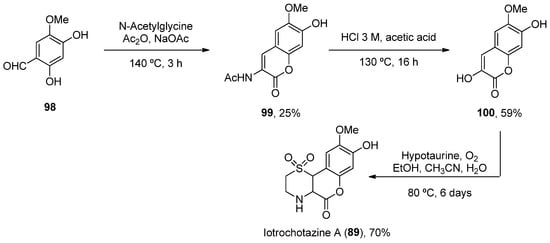
Scheme 15.
Synthesis of iotrochotazine A.