Atrial fibrillation (AF) is the most common cardiac arrhythmia in the world, with an estimated prevalence of 2 to 4%. It is defined as supraventricular tachyarrhythmia with uncoordinated atrial electrical activation, which results in chaotic and irregular activation of atrial pacemaker cells and thus ineffective mechanical contraction of the atrial chambers.
- alarmins
- S100 protein
- HMGB1
- heat shock proteins
1. Introduction
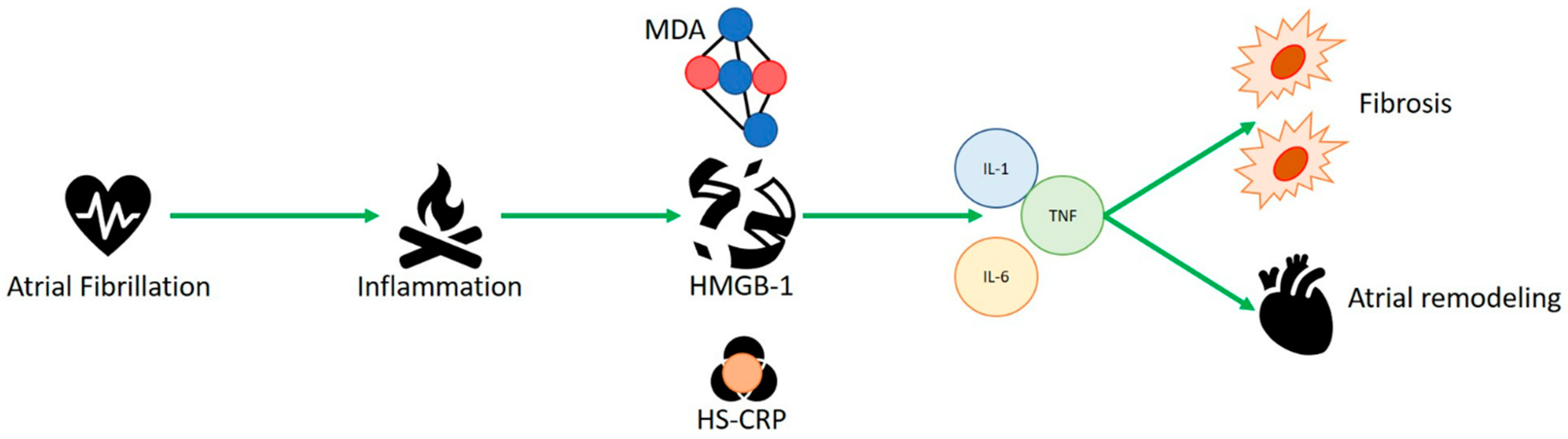
Advanced age is one of the independent risk factors, correlated with multiple disorders such as diabetes, hypertension, heart failure, obesity, and obstructive sleep apnea syndrome (OSAS) [3][1]. Because of the aging of the global population, it is expected to be more prevalent in individuals aged over 65 by 2060 [4,5][2][3]. The interaction of distinct factors, such as inflammation, structural remodeling, fibrosis, and ion-channel dysfunction, guarantees the onset of a complex pathophysiological mechanism that starts atrial fibrillation and worsens structural and electrical changes in the atria [2][4]. The generation of quick multiple ectopic electrical pulses is able to start and sustain irregular electrical activity of atrial fibrillation. The most common locations of occurrence of ectopic foci are the pulmonary veins, whose isolation is the cornerstone of catheter ablation procedures [6[5][6],7], and less commonly the interatrial septum, coronary sinus, and superior vena cava [8,9][7][8]. These electrical alterations also help the AF-associated hypercoagulable state. Failed electrical regularity and excessive ectopic contractility antagonize local atrial hypo contractility, increasing endothelial expression of plasminogen activator inhibitor (PAI-1) [10][9], contributing to clot generation. Various diseases are risk factors for the occurrence of AF, such as hypertension [11][10], heart failure [12][11] and diabetes [13,14][12][13]. Their common factor is represented by the inflammatory state involved in each of these disorders, which plays a key role in the pathophysiology of AF by mediating the production of cytokines and reactive oxygen species that can increase the disease state, fibrosis, and atrial remodeling [15][14]. Hypo contractility, as well as blood stasis, promotes the development of endothelial microdamage, which attracts the migration and infiltration of several cells of the innate immune system, including macrophages and leukocytes. This mechanism damages the atrial architecture, promoting an inflammatory process, remodels the walls of the atrium, fostering fibrous tissue growth and destroying cardiomyocytes, thus increasing the local inflammatory response and helping the expression of endothelial adhesion molecules and inflammatory cytokines [16,17][15][16]. Alarmins represent a group of endogenous molecules characterized by multiple functions. They can be classified into three categories: (1) granule-derived, as α- and β-defensins, cathelicidin (LL37/cathelicidin-related antimicrobial peptide (CRAMP), eosinophil-derived neurotoxin (EDN) and granulysin; (2) nuclear form, including HMGB-1, HMGN1, IL-33, and IL-1α; (3) cytoplasmic, as heat shock proteins (HSP-60, -70, -90, and -96), S100 proteins, ATP and uric acid [18][17]. These intracellular proteins are generally released as inflammatory signal mediators and represent the first defense against infections, as well as during trauma and various metabolic, physical or chemical injuries [19,20][18][19]. Their release can attract additional inflammatory molecules, such as leukocytes, triggering a massive immune local response [21][20] and activating dendritic cells. Roh et al. [22][21] reported that alarmins and various damage-associated molecular patterns (DAMPS), such as HMGB1, S100 and HSP-70, play a key role in the pathogenesis of inflammatory diseases. Several studies have shown a key role for HMGB1, heat shock proteins and s100 proteins in AF physiopathology.
2. HMGB-1 and Human Studies
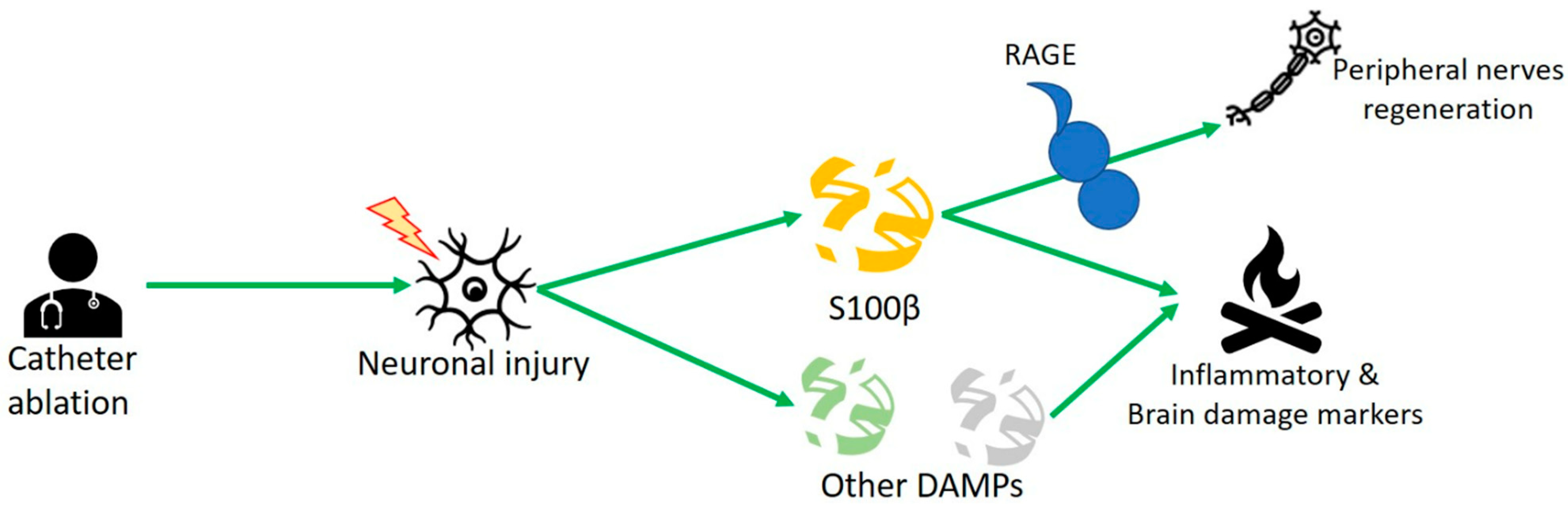
3. S100 Protein and Human Studies
References
- Hart, R.G.; Pearce, L.; Aguilar, M.I. Meta-analysis: Antithrombotic Therapy to Prevent Stroke in Patients Who Have Nonvalvular Atrial Fibrillation. Ann. Intern. Med. 2007, 146, 857–867.
- Colilla, S.; Crow, A.; Petkun, W.; Singer, D.E.; Simon, T.; Liu, X. Estimates of Current and Future Incidence and Prevalence of Atrial Fibrillation in the U.S. Adult Population. Am. J. Cardiol. 2013, 112, 1142–1147.
- Krijthe, B.P.; Kunst, A.; Benjamin, E.; Lip, G.Y.; Franco, O.; Hofman, A.; Witteman, J.C.; Stricker, B.H.; Heeringa, J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013, 34, 2746–2751.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498, Erratum in: Eur. Heart J. 2021, 42, 507.
- Lau, D.H.; Linz, D.; Schotten, U.; Mahajan, R.; Sanders, P.; Kalman, J.M. Pathophysiology of Paroxysmal and Persistent Atrial Fibrillation: Rotors, Foci and Fibrosis. Heart Lung Circ. 2017, 26, 887–893.
- Lau, D.H.; Linz, D.; Sanders, P. New Findings in Atrial Fibrillation Mechanisms. Card Electrophysiol. Clin. 2019, 11, 563–571.
- European Heart Rhythm Association (EHRA); European Cardiac Arrhythmia Scoiety (ECAS); American College of Cardiology (ACC); American Heart Association (AHA); Society of Thoracic Surgeons (STS); Calkins, H.; Brugada, J.; Packer, D.L.; Cappato, R.; Chen, S.A.; et al. HRS/EHRA/ECAS expert Consensus Statement on catheter and surgical ablation of atrial fibrillation: Recommendations for personnel, policy, procedures and follow-up. A report of the Heart Rhythm Society (HRS) Task Force on catheter and surgical ablation of atrial fibrillation. Heart Rhythm. 2007, 4, 816–861.
- Bhatt, H.V.; Fischer, G.W. Atrial Fibrillation: Pathophysiology and Therapeutic Options. J. Cardiothorac. Vasc. Anesthesia 2015, 29, 1333–1340.
- Li, Q.; Lai, Y.; Gao, X.; Li, X.; Deng, C.-Y.; Guo, H.; Zhao, J.; Yang, H.; Xu, Y.; Wu, S.; et al. Involvement of plasminogen activator inhibitor-1 and its related molecules in atrial fibrosis in patients with atrial fibrillation. PeerJ 2021, 9, e11488.
- Yagi, S.; Akaike, M.; Aihara, K.-I.; Ishikawa, K.; Iwase, T.; Ikeda, Y.; Soeki, T.; Yoshida, S.; Sumitomo-Ueda, Y.; Matsumoto, T.; et al. Endothelial Nitric Oxide Synthase–Independent Protective Action of Statin Against Angiotensin II–Induced Atrial Remodeling via Reduced Oxidant Injury. Hypertension 2010, 55, 918–923.
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of Inflammation in Atrial Fibrillation Pathophysiology and Management. Circ. J. 2015, 79, 495–502.
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card. Electrophysiol. Clin. 2021, 13, 25–35.
- Huxley, R.R.; Filion, K.B.; Konety, S.; Alonso, A. Meta-Analysis of Cohort and Case–Control Studies of Type 2 Diabetes Mellitus and Risk of Atrial Fibrillation. Am. J. Cardiol. 2011, 108, 56–62.
- Saito, S.; Teshima, Y.; Fukui, A.; Kondo, H.; Nishio, S.; Nakagawa, M.; Saikawa, T.; Takahashi, N. Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc. Res. 2014, 104, 5–14.
- Chen, G.; Chelu, M.G.; Dobrev, D.; Li, N. Cardiomyocyte Inflammasome Signaling in Cardiomyopathies and Atrial Fibrillation: Mechanisms and Potential Therapeutic Implications. Front. Physiol. 2018, 9, 1115.
- Van Linthout, S.; Miteva, K.; Tscho¨pe, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014, 102, 25.
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol Rev. 2017, 280, 41–56.
- Rider, P.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, I. Alarmins: Feel the Stress. J. Immunol. 2017, 198, 1395–1402.
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365.
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837.
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27.
- Casciaro, M.; Gangemi, S.; Imbalzano, E.; Quartuccio, S.; Di Salvo, E.; Crea, T. Association between HMGB1 and asthma: A literature review. Clin. Mol. Allergy 2017, 15, 12.
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849.
- Qu, C.; Wang, X.-W.; Huang, C.; Qiu, F.; Xiang, X.-Y.; Lu, Z.-Q. High mobility group box 1 gene polymorphism is associated with the risk of postoperative atrial fibrillation after coronary artery bypass surgery. J. Cardiothorac. Surg. 2015, 10, 88.
- Echahidi, N.; Pibarot, P.; O’Hara, G.; Mathieu, P. Mechanisms, Prevention, and Treatment of Atrial Fibrillation After Cardiac Surgery. J. Am. Coll. Cardiol. 2008, 51, 793–801.
- Xu, Q.; Bo, L.; Hu, J.; Geng, J.; Chen, Y.; Li, X.; Chen, F.; Song, J. High mobility group box 1 was associated with thrombosis in patients with atrial fibrillation. Medicine 2018, 97, e0132.
- Gurses, K.M.; Kocyigit, D.; Yalcin, M.U.; Canpinar, H.; Evranos, B.; Canpolat, U.; Yorgun, H.; Sahiner, L.; Guc, D.; Aytemir, K. Platelet Toll-like receptor and its ligand HMGB-1 expression is increased in the left atrium of atrial fibrillation patients. Cytokine 2018, 103, 50–56.
- Migdady, I.; Russman, A.; Buletko, A.B. Atrial Fibrillation and Ischemic Stroke: A Clinical Review. Semin. Neurol. 2021, 41, 348–364.
- Wu, Y.; Zhang, K.; Zhao, L.; Guo, J.; Hu, X.; Chen, Z. Increased serum HMGB1 is related to oxidative stress in patients with atrial fibrillation. J. Int. Med. Res. 2013, 41, 1796–1802.
- Dumitriu, I.E.; Baruah, P.; Manfredi, A.; E Bianchi, M.; Rovere-Querini, P. HMGB1: An immmune odyssey. Discov. Med. 2005, 5, 388–392.
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57.
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 1867, 118677.
- Kozlyuk, N.; Monteith, A.J.; Garcia, V.; Damo, S.M.; Skaar, E.P.; Chazin, W.J. S100 Proteins in the Innate Immune Response to Pathogens. Adv. Struct. Saf. Stud. 2019, 1929, 275–290.
- Scherschel, K.; Hedenus, K.; Jungen, C.; Lemoine, M.D.; Rübsamen, N.; Veldkamp, M.W.; Klatt, N.; Lindner, D.; Westermann, D.; Casini, S.; et al. Cardiac glial cells release neurotrophic S100B upon catheter-based treatment of atrial fibrillation. Sci. Transl. Med. 2019, 11, eaav7770.
- Kato, T.; Sekiguchi, A.; Sagara, K.; Tanabe, H.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.-T.; Yamashita, T. Endothelial–mesenchymal transition in human atrial fibrillation. J. Cardiol. 2016, 69, 706–711.
- Scherschel, K.; Hedenus, K.; Jungen, C.; Münkler, P.; Willems, S.; Anwar, O.; Klatt, N.; Eickholt, C.; Meyer, C. Impact of the ablation technique on release of the neuronal injury marker S100B during pulmonary vein isolation. EP Eur. 2020, 22, 1502–1508.
- Galenko, O.; Jacobs, V.; Knight, S.; Bride, D.; Cutler, M.J.; Muhlestein, J.B.; Carlquist, J.L.; Anderson, J.L.; Knowlton, K.U.; Bunch, T.J. Circulating Levels of Biomarkers of Cerebral Injury in Patients with Atrial Fibrillation. Am. J. Cardiol. 2019, 124, 1697–1700.
- Sramko, M.; Peichl, P.; Wichterle, D.; Tintera, J.; Maxian, R.; Weichet, J.; Knesplova, L.; Franekova, J.; Pasnisinova, S.; Kautzner, J. A Novel Biomarker-Based Approach for the Detection of Asymptomatic Brain Injury During Catheter Ablation of Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2013, 25, 349–354.
- Marchi, N.; Rasmussen, P.; Kapural, M.; Fazio, V.; Kight, K.; Mayberg, M.R.; Kanner, A.; Ayumar, B.; Albensi, B.; Cavaglia, M.; et al. Peripheral markers of brain damage and blood-brain barrier dysfunction. Restor. Neurol. Neurosci. 2003, 21, 109–121.