Parkinson’s disease (PD) is a devastating neurodegenerative disease characterized by progressive destruction of dopaminergic tissue in the central nervous system (CNS). There is no cure for the disease, with current pharmacological treatments aimed at controlling the symptoms. Therefore, there is an unmet need for new treatments for PD. In addition to new therapeutic options, there exists the need for improved efficiency of the existing ones, as many agents have difficulties in crossing the blood–brain barrier (BBB) to achieve therapeutic levels in the CNS or exhibit inappropriate pharmacokinetic profiles, thereby limiting their clinical benefits. To overcome these limitations, an interesting approach is the use of drug delivery systems, such as polymeric microparticles (MPs) that allow for the controlled release of the active ingredients targeting to the desired site of action, increasing the bioavailability and efficacy of treatments, as well as reducing the number of administrations and adverse effects.
Parkinson’s disease (PD) is a devastating neurodegenerative disease characterized by progressive destruction of dopaminergic tissue in the central nervous system (CNS). There is no cure for the disease, with pharmacological treatments aimed at controlling the symptoms. Therefore, there is an unmet need for new treatments for PD. In addition to new therapeutic options, there exists the need for improved efficiency of the existing ones, as many agents have difficulties in crossing the blood–brain barrier (BBB) to achieve therapeutic levels in the CNS or exhibit inappropriate pharmacokinetic profiles, thereby limiting their clinical benefits. To overcome these limitations, an interesting approach is the use of drug delivery systems, such as polymeric microparticles (MPs) that allow for the controlled release of the active ingredients targeting to the desired site of action, increasing the bioavailability and efficacy of treatments, as well as reducing the number of administrations and adverse effects.
- Parkinson’s disease
- polymeric microparticles
- polymeric nanoparticles
1. Introduction
2. Investigational Polymeric Microparticles for the Treatment of Parkinson’s Disease
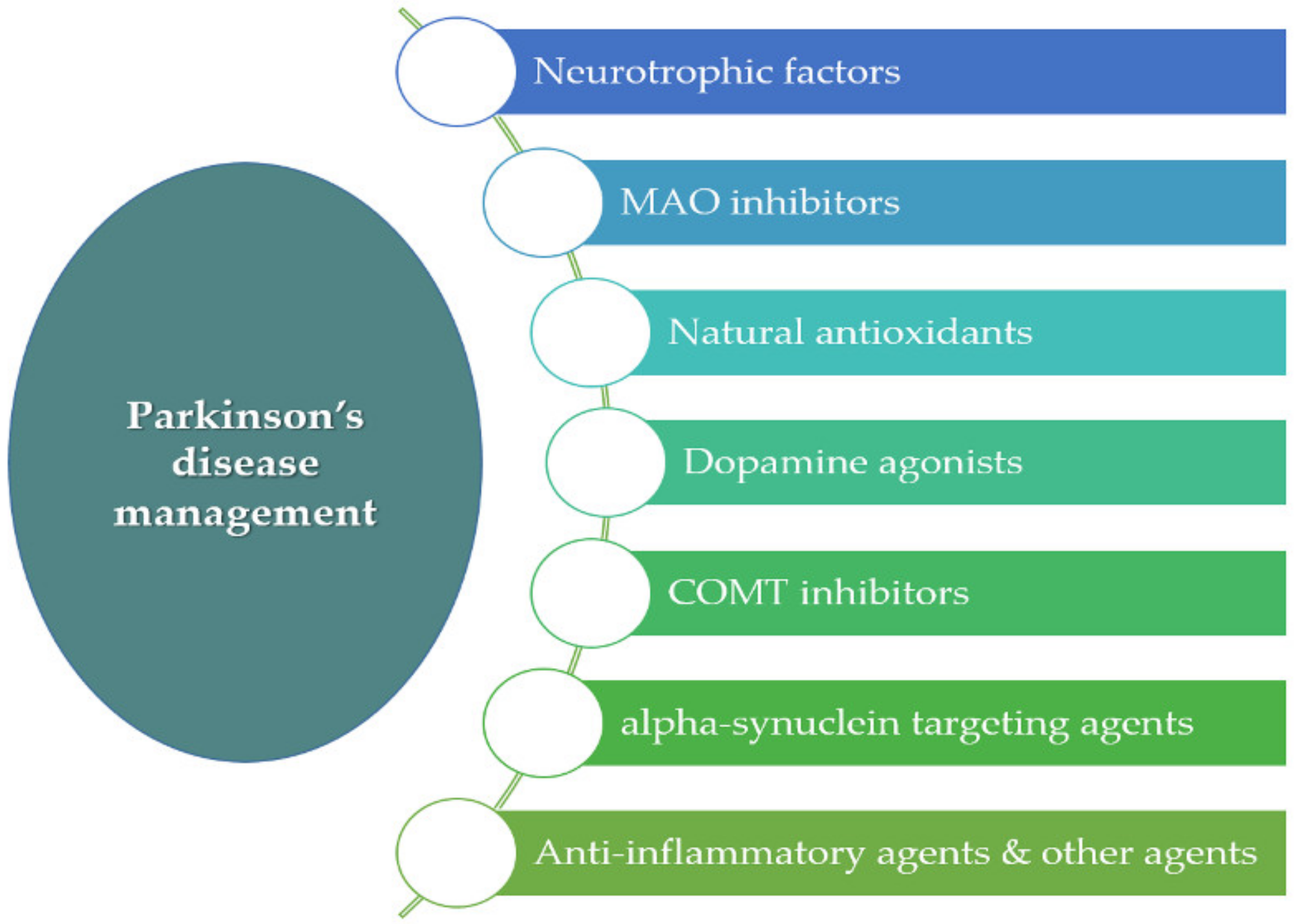
In recent years, different multiparticulate systems, such as polymeric MPs, have been designed as potential therapeutic approaches for PD. MPs are structures with sizes ranging from 1 to 1000 µm [26]. Among the advantages of MPs is the possibility of achieving the controlled release of the active ingredients at the target site. This is of great interest for drugs that have limited access to the CNS. In addition, reduced adverse systemic effects and less frequent dosing intervals can be also achieved by MPs. Under investigation are different antiparkinsonian agents encapsulated within polymeric MPs, including neurotrophic factors such as glial-cell-derived neurotrophic factor (GDNF) and vascular endothelial growth factor (VEGF), antioxidant agents, inhibitors of different enzymes such as monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT), anti-inflammatory compounds, as well as α-synuclein-targeting agents.2.1. Neurotrophic Factors
Neurotrophic factors (NTFs) are biological molecules that influence several neuronal functions, including cell survival and axonal growth [27]. For instance, GDNF has demonstrated to be able to protect and promote neuronal growth, also improving motor functions in animal models of PD [27]. However, as the BBB is a major entry restriction, its high molecular weight and water solubility hinders the access of GDNF to the brain parenchyma. Thus, one viable route of administration is by means of viral vectors integrating the GDNF gene, or by implanting cells, semipermeable structures, or grafts that secrete the neurotrophic factor [28]. In addition, GDNF exhibits a short biological elimination half-life due to its unstable nature. For these reasons, encapsulating this neurotrophic factor within polymeric microparticulate systems may be an interesting approach for brain delivery, as these systems can protect GDNF against external factors, also providing sustained release [29]. The use of GDNF has been Investigated in combination with cell transplants, although this strategy does not lead to direct cell growth, which is necessary for full functional recovery. Furthermore, GDNF must be released transiently at low doses rather than in a constant manner over several months, since continuous release may affect synaptic integration of the transplanted and host tissues. Therefore, given the complex mechanism associated with PD, an interesting strategy may be the design of devices able to release GDNF in the nigral region to improve immediate transplant survival, and in addition, possibly release another factor, such as BDNF, to stimulate neurite outgrowth in the striatal region [30]. Brain-derived neurotrophic factor (BDNF) stimulates and controls growth of new neurons from neural stem cells (neurogenesis) [31,32,33][31][32][33]. In vitro studies have also provided evidence that BDNF can improve DA production, axonal extension, and survival in cultured dopaminergic neurons [34]. Abnormally low levels of this factor have been associated with neurodegenerative diseases such as PD [35]. BDNF could be used to reduce neurodegeneration and enhance the success of cell transplants required for long-term therapy and potential reversal of disease by promoting reinnervation [36]. However, clinical efficacy has been severely limited by its limited access to the CNS [28]. In human traumatic brain injury (TBI), day-of-injury serum BDNF has been associated with TBI diagnosis also providing 6-month prognostic information regarding recovery from the injury [37]. Vascular endothelial growth factor (VEGF) is an angiogenic factor with specificity for endothelial cells that has demonstrated neuroprotective effects in animal models of PD [38]. However, the use of VEGF as a neuroprotective factor is complicated, as its effect is dose-dependent, with high levels of VEGF inducing brain edema [39]. As with other NFs, clinical use is limited by its rapid degradation and difficulty in crossing the BBB [40]. To obtain a continuous and direct release of VEGF to the CNS, different intracranial administration strategies, such as MP administration, have been tested in animal models of PD [41] (Figure 4).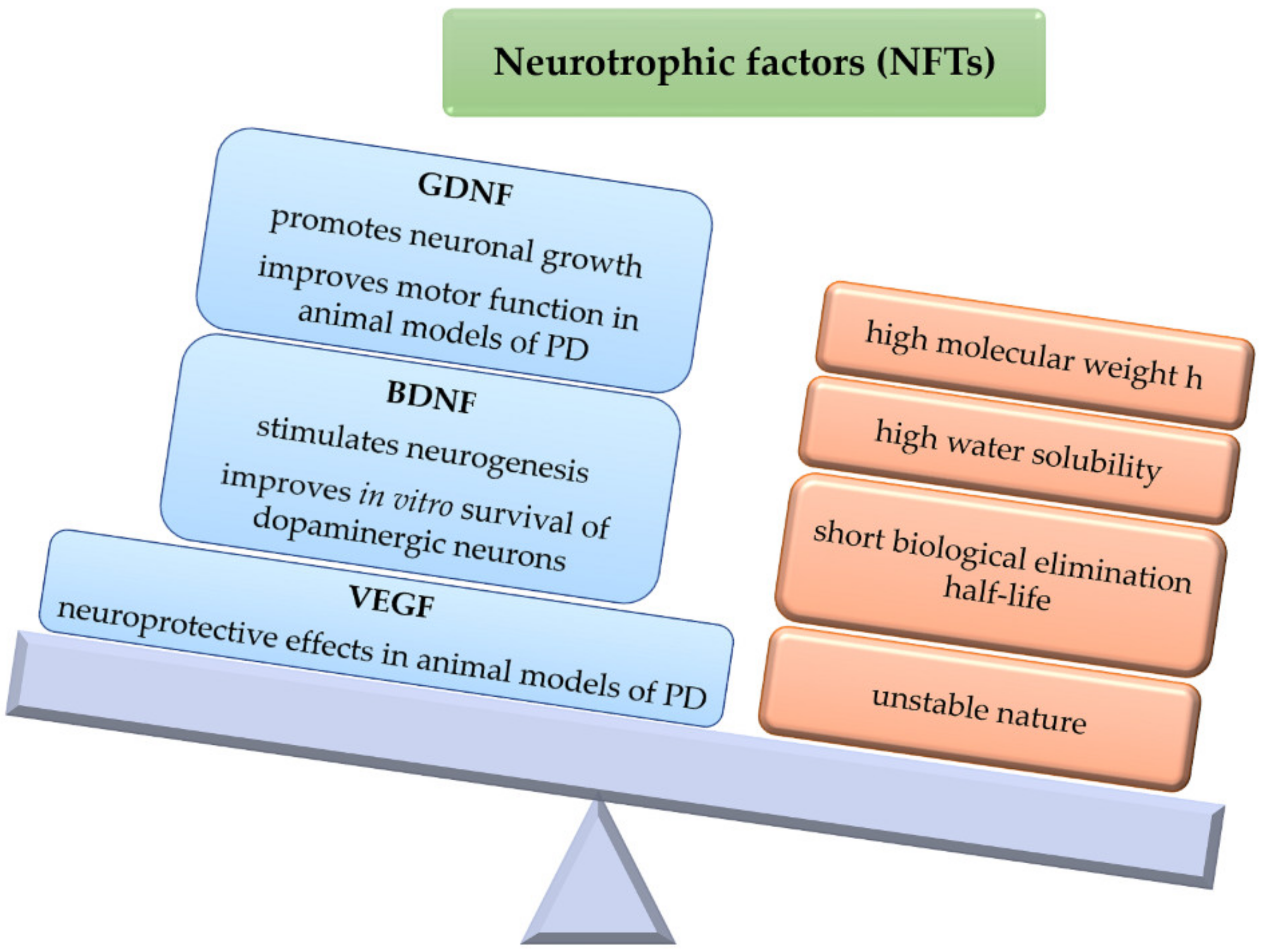
2.2. MAO Inhibitors/Antioxidants
Oxidative stress is a result of various metabolic activities which are essential for life and usually leads to the formation of reactive oxygen species (ROS) and reactive nitrogen species (RNS). It has been associated with the development of PD [50], as increased oxidative stress has been related to the overexpression of α-syn aggregates [51]. The mechanisms involved in neuronal degeneration occurring in PD are complex and remain to be fully elucidated, although it is known that the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) is responsible for the characteristic motor symptoms present in the disease [1,2][1][2]. Accumulating evidence suggests that oxidative damage and mitochondrial dysfunction contribute to the cascade of events leading to degeneration of dopaminergic neurons [52,53,54][52][53][54]. This is supported by post-mortem brain analyses showing increased levels of 4-hydroxyl-2-nonenal (HNE), a by-product of lipid peroxidation [55], the formation of DNA and RNA oxidation products (8-hydroxy-deoxyguanosine and 8-hydroxy-guanosine) [56[56][57],57], and carbonyl modifications of soluble proteins [58]. The link between oxidative stress and dopaminergic neuronal degeneration is further supported by modelling the motor aspects of PD in animal models using toxins that cause oxidative stress, including MPTP, rotenone (RT), 6-OHDA, and 1,1′-dimethyl-4,4′-bipyridinium dichloride (paraquat) [59,60,61][59][60][61]. For this reason and given that oxidative imbalance in PD has a multifactorial origin, the use of antioxidant agents could be a viable therapeutic strategy [62]. In this regard, multiple alternatives have been proposed based on the use of iron chelators, ROS scavengers (vitamins, polyphenols, glutathione, etc.), and other substances (vegetable extracts, melatonin, nicotine, etc.) [62,63][62][63]. In addition, it seems that MAO-B (monoamine oxidase-B) plays an important role in the production of reactive oxygen intermediates (ROI) in activated astrocytes. MAO-B metabolizes the MPTP toxin to 1-methyl-4-phenylpyridium (MPP+), leading to the production of ROI and eventually to cell death [64]. Rasagiline (RG) is a therapeutic agent belonging to the group of MAO-B inhibitors (MAOIs) which has demonstrated neuroprotective activity against MPTP and 6-OHDA animal models of PD [65,66][65][66]. Rasagiline is used for the symptomatic treatment of PD. A recent study conducted in PD patients by Im et al. [67], when investigating the effects of RG on regional cerebral blood flow (rCBF) by single-photon-emission-computed tomography (SPECT), showed that adjunctive RG therapy has beneficial effects on perfusion in the precuneus of PD patients due to its neuroprotective effects. The low oral bioavailability of RG (around 36%) and its short elimination half-life (0.6–2 h) [68] make it a suitable candidate for the design of controlled-release systems. For this, Fernández et al. [69] developed PLGA MPs loaded with RG mesylate (RM). The efficacy of this system was evaluated in male Wistar rats in an RT-induced model of PD. Daily intraperitoneal (i.p.) administration of RT at a dose of 2 mg/kg/day resulted in neuronal degeneration and behavioral deficits resembling those occurring in PD. Treated animals received the same daily dose of RT for 45 days and RM in saline (1 mg/kg/day) or RM-loaded PLGA MPs, which were assayed at two dose levels: high dose (amount of MPs equivalent to 15 mg/kg RM injected every 15 days) and low dose (amount of MPs equivalent to 7.5 mg/kg RM injected every 15 days). The results demonstrated a robust effect of high-dose RM-loaded MPs on all behavioral tests (catalepsy, akinesia, swim test) which resulted in better outcomes than RM given in solution (1 mg/kg/day). Furthermore, Nissl staining of brain sections showed selective degeneration of the substantia nigra (SN) dopaminergic neurons in animals treated with RT, which was markedly reverted by the administration of high-dose RM-loaded PLGA MPs. Interestingly, PET/CT analysis (positron emission tomography/computed tomography) using 18F-DG (fluorodeoxyglucose F18) resulted in mean increases in the radiotracer in striatum and SN of around 40% when RM-loaded PLGA MPs were given to the animals, thereby indicating the efficacy of the microparticulate drug delivery system developed for RM. This study continued in 2012 [70]. In this case, an advanced stage of neurodegeneration was achieved by daily i.p. injections of RT (2 mg/kg). On day 15, animals received RM in saline (1 mg/kg/day) or encapsulated within PLGA microspheres (amount of microspheres equivalent to 15 mg/kg RM given on days 15 and 30). After 45 days RM showed a robust effect on all outcomes evaluated (behavioral tests, Nissl staining, and PCR or Polymerase Chain Reaction), with non-statistically significant differences found between its administration in solution or encapsulated within MPs; however, with the new delivery system, administration of RM could be performed every two weeks instead of daily. Kanwar et al. [22] evaluated the efficacy of RM-loaded polycaprolactone (PCL) MPs in Sprague Dawley rats. For the development of PD-like symptoms, stereotaxic infusion of RT (6 μg/2 μL vehicle) was conducted in the animals. After recovery from surgery, animals received RM in solution (1 mg/kg/day), blank PCL MPs, or RM-loaded PCL MPs (equivalent to 30 mg/kg RM given once a month by subcutaneous (s.c.) injection). Significant differences in behavioral tests (locomotor activity, grip strength) and biochemical markers of oxidative stress (lipid peroxidation, reduced glutathione, etc.) were observed between the RM-treated groups and control animals. Non-statistically significant differences were found when RM was given in solution or encapsulated within PCL MPs, but administration of the microparticulate formulation could reduce the need for frequent dosing intervals, thereby resulting in better patient compliance in the treatment of PD. A few polymeric microparticulate systems have been developed for the encapsulation of MAO-B inhibitors. From the formulation developed, satisfactory results were obtained in animal models of PD, although several issues should be addressed, such as pharmacokinetics and toxicity, as long-term systemic medication commonly leads to deleterious side effects.2.3. Dopamine Agonists
DA receptor agonists may be able to prevent the nigrostriatal dopaminergic cell loss occurring in PD due to their antioxidant and levodopa-sparing effects. For this, pramipexole [71[71][72][73],72,73], apomorphine [74], and ropinirole [75] have been studied in different neurodegenerative animal models, with the outcomes of these studies demonstrating that, in general, these DA agonists are able to prevent the loss of dopaminergic neurons. Furthermore, recent therapeutical approaches for PD are exploring the use of radical scavenging materials, as polymers as catechol structures have demonstrated antioxidant activities due to their physicochemical properties. In this regard, Newland et al. [76] synthesized photocrosslinkable DA-containing poly(β-amino ester) (DPAE) from poly(ethylene glycol) diacrylate (PEGDA) and dopamine hydrochloride using Michael-type addition. The authors developed a water-in-oil emulsion technique to photocrosslink the polymer into spherical MPs. The ability of the microparticulate formulation to capture ROS was analyzed by the 2,2-diphenyl-1-picrylhydrazyl (DPPH) method. DPAE MPs at concentrations of 5250 spheres/mL and 7000 spheres/mL reduced the formation of DPPH radicals up to 47 ± 9% and 56 ± 5%, respectively. In contrast, PEG MPs at a concentration of 7000 spheres/mL led to a reduction of only 5 ± 1%. The free radical activity of DPAE MPs increased in a dose-dependent manner up to 56%, with this level of antioxidant activity being slightly higher than that produced by ascorbic acid (10 μM), a well-known antioxidant. Furthermore, tests conducted in dopaminergic SH-SY5Y cells, primary astrocytes, and primary embryonic rat ventral midbrain cultures showed that the concentrations required for radical scavenging were non-toxic, as reduction in metabolic cell activity or morphological alterations did not occur. Negro et al. [77] developed ropinirole (RP)-loaded PLGA MPs as a controlled delivery system for this DA agonist. The formulation exhibited sustained in vitro release of the drug (78.23 µg/day/10 mg MPs) for 19 days. The efficacy of the new delivery system was evaluated in an RT model of PD induced in male Wistar rats. For this, animals received daily i.p. doses of RT (2 mg/kg). Once PD-like symptoms appeared (day 15), animals were given either RP in saline (1 mg/kg/day for 45 days) or RP-loaded PLGA MPs at two dose levels (amounts of MPs equivalent to 7.5 mg/kg or 15 mg/kg RP given on days 15 and 30, respectively). Behavioral outcomes (akinesia, catalepsy, rotarod, swim test) and brain analyses (Nissl staining, glial fibrillary acidic protein (GFAP), TH immunohistochemistry) showed that animals receiving RP either in solution or encapsulated within the MPs reverted PD-like symptoms, with the best results obtained with the MPs at the highest dose assayed.2.4. COMT Inhibitors
Catechol-O-methyltransferase (COMT) is a selective and widely distributed enzyme involved in the catabolism of levodopa, with tolcapone (TC) being a potent COMT inhibitor both in the brain and peripheral tissues, that can slow down levodopa metabolism, thereby leading to a prolongation of its effect in the treatment of PD. Casanova et al. [78] developed TC-loaded PLGA MPs which exhibited zero-order in vitro release of the drug for 30 days. The new delivery system prepared with 120 mg of TC and 400 mg of PLGA 502 was tested in an RT model of PD induced in male Wistar rats. Daily i.p. injections of the neurotoxin (2 mg/kg) were given to induce neurodegeneration. Once established, animals received TC in saline (3 mg/kg/day) or encapsulated within the MPs (amount of MPs equivalent to 3 mg/kg/day TC every 14 days). Brain analyses of Nissl staining, GFAP, and TH as well as behavioral testing (akinesia, catalepsy, swim test) showed that the new delivery system developed for TC was able to efficiently revert PD-like symptoms in the animal model assayed.2.5. α-Synuclein-Targeting Agents
Alpha-synuclein (α-syn) is a key protein involved in the pathogenesis of PD. The exact function of α-syn remains still largely unknown, although mounting evidence supports the fact that α-syn is involved in synaptic plasticity and neurotransmitter release [79,80][79][80]. Under normal conditions, native α-syn exists in a dynamic equilibrium between unfolded monomers and α-helically folded tetramers, with a low tendency for aggregation [81]. The reduction in the tetramer:monomer ratio and the consequent increase in the level of α-syn unfolded monomers favor its aggregation [82], which involves a conformational change where the protein adopts a β-sheet-rich structure that facilitates its aggregation into oligomers, protofibrils, and insoluble fibrils that finally accumulate in the form of Lewy bodies [83]. Lewy bodies are a characteristic feature of PD, being closely related to its progression [84], as the appearance of Lewy bodies induces alterations in synapses, mitochondrial dysfunction, and deterioration of the endoplasmic reticulum functionality. These circumstances lead to increased metabolic activity, oxidative stress, and protein accumulation, which may explain the subsequent cell death occurring in the disease. Furthermore, misfolded α-syn can spread between different cells and tissues, inducing misfolding of other units of the protein [83]. The most common α-syn-targeting strategies used in the treatment of PD are decreasing the expression of α-syn with antisense oligonucleotides or miRNA, inhibiting its aggregation with small molecules, favoring its clearance via autophagy, and preventing the seeding and prion-like spreading of α-syn [81,85,86][81][85][86]. In this regard, immunotherapy is gaining increased attention for the management of PD, with active immunization being of particular interest, as it allows for prolonged treatments without the need for frequent dosing intervals [87]. Immunotherapeutic approaches targeting α-syn have the advantage of addressing several of these mechanisms, with active and passive vaccination being able to prevent neurodegeneration and reduce α-syn accumulation by promoting clearance via autophagy and microglial cells [88,89,90,91][88][89][90][91]. The ability of the combined delivery of antigen plus rapamycin (RAP) in the form of nanoparticles in inducing antigen-specific regulatory T cells (Tregs) has been already demonstrated [92]. Rockenstein et al. [87] adapted this approach to α-syn by developing an antigen-presenting cell-targeting glucan microparticle (GP) vaccine delivery system. This active immunization system with immunomodulatory activity was composed of GP-MPs loaded with RAP and α-syn as antigen to produce the active humoral immunization (GP+RAP/α-syn). The immunity produced was compared to that of GP-alone, GP-α-syn, and GP+RAP in transgenic (tg) PDGFβ α-syn Line D male and female mice. The effect was analyzed using neuropathological and biochemical markers. Mice treated with GP-RAP and GP-RAP/α-syn showed an increase in the number of Treg lymphocytes in the CNS, decreased levels of proinflammatory cytokines (IL-6 and TNF-α), and increased levels of TGF-β1 (transforming growth factor-beta 1). Animals treated with GP-α-syn and GP-RAP/α-syn presented significant increases in anti-α-syn antibody titers, decreased α-syn aggregates and neurodegeneration, and increased functional recovery markers of neurodegeneration (Arc protein and TH). Therefore, combined therapy (RAP and the antigen) led to better responses than monotherapies, possibly due to the combination of the immunomodulatory activity of RAP and immunization by α-syn. The authors concluded that this new vaccine delivery system, which induced both regulatory Tregs and anti-α-synuclein antibody titers, demonstrated a satisfactory effect in reducing α-syn accumulation, neurodegeneration, and inflammation. Authors claimed that this combined vaccine may be more potent than conventional immunization, which is only based on cellular or humoral immunization, with the potential to be further investigated in synucleinopathies, such as PD and dementia with Lewy bodies, among others. From the promising results obtained, further research should be conducted to determine if combining humoral and cellular immunization might synergistically reduce inflammation and improve microglial-mediated α-syn clearance. Table 1 summarizes the polymeric microparticulate systems under investigation as potential new therapeutic approaches for PD.| Category | Active Compound | Polymer | Research Model | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Neurotrophic factors | GDNF | PLGA shell/Collagen core | Cell culture: neural stem/progenitor cells. | [44] | ||||
| GDNF/BDNF | PLGA/PEG | Cell cultures: brain/glial cells, microglia, and astrocytes. Animal study: female Sprague Dawley rats. |
[43] | |||||
| GDNF | PLGA | Animal study: partial and progressive 6-OHDA-induced model of PD. Female Sprague Dawley rats. |
[42] | |||||
| GDNF | PLGA | Animal study: 6-OHDA-induced model of PD. Female Sprague Dawley rats. | [46] | |||||
| GDNF | PLGA | Animal study: MPTP-induced model of PD. | Macaca fascicularis | . | [47] | |||
| GDNF/VEGF | PLGA | Animal study: 6-OHDA-induced model of PD. Female albino Sprague Dawley rats. | [40] | |||||
| MAO inhibitors/Antioxidants |
Rasagiline | PLGA | SKN-AS cell culture, H | 2 | O | 2 | neurotoxin. Animal study: RT-induced model of PD. Male Wistar rats. |
[69] |
| Rasagiline | PLGA | SKN-AS cell culture, H | 2 | O | 2 | neurotoxin. Animal study: advanced RT model of PD. Male Wistar rats. |
[70] | |
| Rasagiline | Polycaprolactone | Animal study: RT-induced model of PD. Male Sprague Dawley rats. | [22] | |||||
| Dopamine agonists |
Dopamine | Poly(β-aminoester) | Cell cultures: SH-SY5Y cells, rat primary astrocytes, embryonic midbrain cultures. | [76] | ||||
| Ropinirole | PLGA | SKN-AS cell culture, H | 2 | O | 2 | neurotoxin. Animal study: RT-induced model of PD. Male Wistar rats. |
[77] | |
| COMT inhibitors | Tolcapone | PLGA | Animal study: RT-induced model of PD. Male Wistar rats. | [78] | ||||
| Alpha-synuclein-targeting agents | α-syn + Rapamycin | Glucan | Animal study: transgenic PDGFβ α-syn. Male and female Line D mice. | [87] |
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118.
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953.
- World Health Organization. Parkinson Disease: A Public Health Approach: Technical Brief; World Health Organization: Geneva, Switzerland, 2022.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Mahlknecht, P.; Seppi, K.; Poewe, W. The concept of prodromal Parkinson’s disease. J. Park. Dis. 2015, 5, 681–697.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548–560.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12.
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995.
- Draoui, A.; El Hiba, O.; Aimrane, A.; El Khiat, A.; Gamrani, H. Parkinson’s disease: From bench to bedside. Rev. Neurol. 2020, 176, 543–559.
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350.
- Gárdián, G.; Vécsei, L. Medical treatment of Parkinson’s disease: Today and the future. Int. J. Clin. Pharm. 2010, 48, 633–642.
- Liao, X.; Wu, N.; Liu, D.; Shuai, B.; Li, S.; Li, K. Levodopa/carbidopa/entacapone for the treatment of early Parkinson’s disease: A meta-analysis. Neurol Sci. 2020, 41, 2045–2054.
- Rogers, G.; Davies, D.; Pink, J.; Cooper, P. Parkinson’s disease: Summary of updated NICE guidance. BMJ 2017, 358, j1951.
- Hernando, S.; Gartziandia, O.; Herran, E.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M. Advances in nanomedicine for the treatment of Alzheimer’s and Parkinson’s diseases. Nanomedicine 2016, 11, 1267–1285.
- Barcia, E.; Boeva, L.; García-García, L.; Slowing, K.; Fernández-Carballido, A.; Casanova, Y.; Negro, S. Nanotechnology-based drug delivery of ropinirole for Parkinson’s disease. Drug Deliv. 2017, 24, 1112–1123.
- Lengyel, M.; Kállai-Szabó, N.; Antal, V.; Laki, A.J.; Antal, I. Microparticles, microspheres, and microcapsules for advanced drug delivery. Sci. Pharm. 2019, 87, 20.
- Baskin, J.; Jeon, J.E.; Lewis, S.J.G. Nanoparticles for drug delivery in Parkinson’s disease. J. Neurol. 2021, 268, 1981–1994.
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387.
- Svenson, S.; Wolfgang, M.; Hwang, J.; Ryan, J.; Eliasof, S. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J. Control. Release 2011, 153, 49–55.
- Kanwar, N.; Bhandari, R.; Kuhad, A.; Sinha, V.R. Polycaprolactone-based neurotherapeutic delivery of rasagiline targeting behavioral and biochemical deficits in Parkinson’s disease. Drug Deliv. Transl. Res. 2019, 9, 891–905.
- Mittal, D.; Md, S.; Hasan, Q.; Fazil, M.; Ali, A.; Baboota, S.; Ali, J. Brain targeted nanoparticulate drug delivery system of rasagiline via intranasal route. Drug Deliv. 2016, 23, 130–139.
- Alconcel, S.N.S.; Baas, A.S.; Maynard, H.D. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448.
- Kurakhmaeva, K.B.; Djindjikhashvili, I.A.; Petrov, V.E.; Balabanyan, V.U.; Voronina, T.A.; Trofimov, S.S.; Kreuter, J.; Gelperina, S.; Begley, D.; Alyautdin, R.N. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target. 2009, 17, 564–574.
- Bale, S.; Khurana, A.; Reddy, A.S.; Singh, M.; Godugu, C. Overview on therapeutic applications of microparticulate drug delivery systems. Crit. Rev. Drug Carr. Syst. 2016, 33, 309–361.
- Rangasamy, S.B.; Soderstrom, K.; Bakay, R.A.; Kordower, J.H. Neurotrophic factor therapy for Parkinson’s disease. Prog. Brain Res. 2010, 184, 237–264.
- Axelsen, T.M.; Woldbye, D.P.D. Gene Therapy for Parkinson’s Disease, An Update. J. Park. Dis. 2018, 8, 195–215.
- Khalin, I.; Alyautdin, R.; Wong, T.W.; Gnanou, J.; Kocherga, G.; Kreuter, J. Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv. 2016, 23, 3520–3528.
- Zawada, W.M.; Zastrow, D.J.; Clarkson, E.D.; Adams, F.S.; Bell, K.P.; Freed, C.R. Growth factors improve immediate survival of embryonic dopamine neurons after transplantation into rats. Brain Res. 1998, 786, 96–103.
- Zigova, T.; Pencea, V.; Wiegand, S.J.; Luskin, M.B. Intraventricular administration of BDNF increases the number of newly generated neurons in the adult olfactory bulb. Mol. Cell Neurosci. 1998, 11, 234–245.
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J. Neurosci. 2001, 21, 6706–6717.
- Benraiss, A.; Chmielnicki, E.; Lerner, K.; Roh, D.; Goldman, S.A. Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J. Neurosci. 2001, 21, 6718–6731.
- Østergaard, K.; Jones, S.A.; Hyman, C.; Zimmer, J. Effects of donor age and brain-derived neurotrophic factor on the survival of dopaminergic neurons and axonal growth in postnatal rat nigrostriatal cocultures. Exp. Neurol. 1996, 142, 340–350.
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124.
- Yurek, D.M.; Lu, W.; Hipkens, S.; Wiegand, S.J. BDNF enhances the functional reinnervation of the striatum by grafted fetal dopamine neurons. Exp. Neurol. 1996, 137, 105–118.
- Korley, F.K.; Diaz-Arrastia, R.; Wu, A.H.B.; Yue, J.K.; Manley, G.T.; Sair, H.I.; Van Eyk, J.; Everett, A.D.; TRACK-TBI Investigators; Okonkwo, D.O.; et al. Circulating brain-derived neurotrophic factor has diagnostic and prognostic value in traumatic brain injury. J. Neurotrauma 2016, 33, 215–225.
- Yasuhara, T.; Shingo, T.; Kobayashi, K.; Takeuchi, A.; Yano, A.; Muraoka, K.; Matsui, T.; Miyoshi, Y.; Hamada, H.; Date, I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2004, 19, 1494–1504.
- Yasuhara, T.; Shingo, T.; Muraoka, K.; wen Ji, Y.; Kameda, M.; Takeuchi, A.; Yano, A.; Nishio, S.; Matsui, T.; Miyoshi, Y.; et al. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res. 2005, 1038, 1–10.
- Herrán, E.; Ruiz-Ortega, J.Á.; Aristieta, A.; Igartua, M.; Requejo, C.; Lafuente, J.V.; Ugedo, L.; Pedraz, J.L.; Hernández, R.M. In vivo administration of VEGF- and GDNF-releasing biodegradable polymeric microspheres in a severe lesion model of Parkinson’s disease. Eur. J. Pharm. Biopharm. 2013, 85, 1183–1190.
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Del. Rev. 2012, 64, 640–665.
- Jollivet, C.; Aubert-Pouessel, A.; Clavreul, A.; Venier-Julienne, M.C.; Remy, S.; Montero-Menei, C.N.; Benoit, J.P.; Menei, P. Striatal implantation of GDNF releasing biodegradable microspheres promotes recovery of motor function in a partial model of Parkinson’s disease. Biomaterials 2004, 25, 933–942.
- Lampe, K.J.; Kern, D.S.; Mahoney, M.J.; Bjugstad, K.B. The administration of BDNF and GDNF to the brain via PLGA microparticles patterned within a degradable PEG-based hydrogel: Protein distribution and the glial response. J. Biomed. Mater. Res. A 2011, 96, 595–607.
- Gujral, C.; Minagawa, Y.; Fujimoto, K.; Kitano, H.; Nakaji-Hirabayashi, T. Biodegradable microparticles for strictly regulating the release of neurotrophic factors. J. Control. Release 2013, 168, 307–316.
- Gouhier, C.; Chalon, S.; Aubert-Pouessel, A.; Venier-Julienne, M.C.; Jollivet, C.; Benoit, J.P.; Guilloteau, D. Protection of dopaminergic nigrostriatal afferents by GDNF delivered by microspheres in a rodent model of Parkinson’s disease. Synapse 2002, 44, 124–131.
- Garbayo, E.; Montero-Menei, C.N.; Ansorena, E.; Lanciego, J.L.; Aymerich, M.S.; Blanco-Prieto, M.J. Effective GDNF brain delivery using microspheres--a promising strategy for Parkinson’s disease. J. Control. Release 2009, 135, 119–126.
- Garbayo, E.; Ansorena, E.; Lana, H.; Carmona-Abellan, M.D.; Marcilla, I.; Lanciego, J.L.; Luquin, M.R.; Blanco-Prieto, M.J. Brain delivery of microencapsulated GDNF induces functional and structural recovery in parkinsonian monkeys. Biomaterials 2016, 110, 11–23.
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue. Res. 2004, 318, 215–224.
- Casanova, Y.; Negro, S.; Barcia, E. Application of neurotoxin- and pesticide-induced animal models of Parkinson’s disease in the evaluation of new drug delivery systems. Acta Pharm. 2022, 72, 35–58.
- Percário, S.; da Silva Barbosa, A.; Varela, E.L.P.; Gomes, A.R.Q.; Ferreira, M.E.S.; de Nazaré Araújo Moreira, T.; Dolabela, M.F. Oxidatives Stress in Parkinson’s Disease: Potential benefits of antioxidant supplementation. Oxid. Med. Cell Longev. 2020, 2020, 2360872.
- Luk, K.C. Oxidative stress and α-synuclein conspire in vulnerable neurons to promote Parkinson’s disease progression. J. Clin. Investig. 2019, 129, 3530–3531.
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055.
- Zhu, J.; Chu, C.T. Mitochondrial dysfunction in Parkinson’s disease. J. Alzheimer’s Dis. 2010, 20, S325–S334.
- Jenner, P.; Olanow, C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology 2006, 66, S24–S36.
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S36, discussion S28–S36.
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203.
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429.
- Floor, E.; Wetzel, M.G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998, 70, 268–275.
- Richardson, J.R.; Quan, Y.; Sherer, T.B.; Greenamyre, J.T.; Miller, G.W. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol. Sci. 2005, 88, 193–201.
- Callio, J.; Oury, T.D.; Chu, C.T. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J. Biol. Chem. 2005, 280, 18536–18542.
- Perier, C.; Bové, J.; Vila, M.; Przedborski, S. The rotenone model of Parkinson’s disease. Trends Neurosci. 2003, 26, 345–346.
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522.
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. Biometals 2018, 31, 737–747.
- Mandel, S.; Grünblatt, E.; Riederer, P.; Gerlach, M.; Levites, Y.; Youdim, M.B. Neuroprotective strategies in Parkinson’s disease: An update on progress. CNS Drugs 2003, 17, 729–762.
- Cronin, A.; Grealy, M. Neuroprotective and Neuro-restorative Effects of minocycline and rasagiline in a zebrafish 6-hydroxydopamine model of Parkinson’s disease. Neuroscience 2017, 367, 34–46.
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J. Neural. Transm. 2020, 127, 131–147.
- Im, J.J.; Jeong, H.; Chung, Y.A.; Park, J.S.; Heo, Y.; Oh, J.K.; Song, I.U. Neuroprotective effects of rasagiline in Parkinson’s disease: A regional cerebral blood flow study. J. Neuroimaging 2019, 29, 707–711.
- Chen, J.J.; Swope, D.M.; Dashtipour, K. Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin. Ther. 2007, 29, 1825–1849.
- Fernández, M.; Negro, S.; Slowing, K.; Fernández-Carballido, A.; Barcia, E. An effective novel delivery strategy of rasagiline for Parkinson’s disease. Int. J. Pharm. 2011, 419, 271–280.
- Fernández, M.; Barcia, E.; Fernández-Carballido, A.; Garcia, L.; Slowing, K.; Negro, S. Controlled release of rasagiline mesylate promotes neuroprotection in a rotenone-induced advanced model of Parkinson’s disease. Int. J. Pharm. 2012, 438, 266–278.
- Vu, T.Q.; Ling, Z.D.; Ma, S.Y.; Robie, H.C.; Tong, C.W.; Chen, E.Y.; Lipton, J.W.; Carvey, P.M. Pramipexole attenuates the dopaminergic cell loss induced by intraventricular 6-hydroxydopamine. J. Neural Transm. 2000, 107, 159–176.
- Zou, L.; Xu, J.; Jankovic, J.; He, Y.; Appel, S.H.; Le, W. Pramipexole inhibits lipid peroxidation and reduces injury in the substantia nigra induced by the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 mice. Neurosci. Lett. 2000, 281, 167–170.
- Iravani, M.M.; Haddon, C.O.; Cooper, J.M.; Jenner, P.; Schapira, A.H. Pramipexole protects against MPTP toxicity in non-human primates. J. Neurochem. 2006, 96, 1315–1321.
- Kyriazis, M. Neuroprotective, anti-apoptotic effects of apomorphine. J. Anti-Aging Med. 2003, 6, 21–28.
- Iida, M.; Miyazaki, I.; Tanaka, K.; Kabuto, H.; Iwata-Ichikawa, E.; Ogawa, N. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 1999, 838, 51–59.
- Newland, B.; Wolff, P.; Zhou, D.; Wang, W.; Zhang, H.; Rosser, A.; Wang, W.; Werner, C. Synthesis of ROS scavenging microspheres from a dopamine containing poly(β-amino ester) for applications for neurodegenerative disorders. Biomater. Sci. 2016, 4, 400–404.
- Negro, S.; Boeva, L.; Slowing, K.; Fernandez-Carballido, A.; Garcia-García, L.; Barcia, E. Efficacy of ropinirole-loaded PLGA microspheres for the reversion of rotenone-induced parkinsonism. Curr. Pharm. Des. 2017, 23, 3423–3431.
- Casanova, Y.; Negro, S.; Slowing, K.; García-García, L.; Fernández-Carballido, A.; Rahmani, M.; Barcia, E. Micro- and nano-systems developed for tolcapone in Parkinson’s disease. Pharmaceutics 2022, 14, 1080.
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568.
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48.
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Müller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating native α-synuclein tetramers in mice causes a L-DOPA-responsive motor syndrome closely resembling Parkinson’s disease. Neuron 2018, 100, 75–90.e75.
- Brás, I.C.; Xylaki, M.; Outeiro, T.F. Chapter 4—Mechanisms of alpha-synuclein toxicity: An update and outlook. In Progress in Brain Research; Björklund, A., Cenci, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 252, pp. 91–129.
- Chatterjee, D.; Kordower, J.H. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104587.
- Valera, E.; Masliah, E. Therapeutic approaches in Parkinson’s disease and related disorders. J. Neurochem. 2016, 139, 346–352.
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235.
- Rockenstein, E.; Ostroff, G.; Dikengil, F.; Rus, F.; Mante, M.; Florio, J.; Adame, A.; Trinh, I.; Kim, C.; Overk, C.; et al. Combined active humoral and cellular immunization approaches for the treatment of synucleinopathies. J. Neurosci. 2018, 38, 1000–1014.
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868.
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE 2011, 6, e19338.
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879.
- Mandler, M.; Valera, E.; Rockenstein, E.; Mante, M.; Weninger, H.; Patrick, C.; Adame, A.; Schmidhuber, S.; Santic, R.; Schneeberger, A.; et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 10.
- Maldonado, R.A.; LaMothe, R.A.; Ferrari, J.D.; Zhang, A.H.; Rossi, R.J.; Kolte, P.N.; Griset, A.P.; O’Neil, C.; Altreuter, D.H.; Browning, E.; et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, E156–E165.