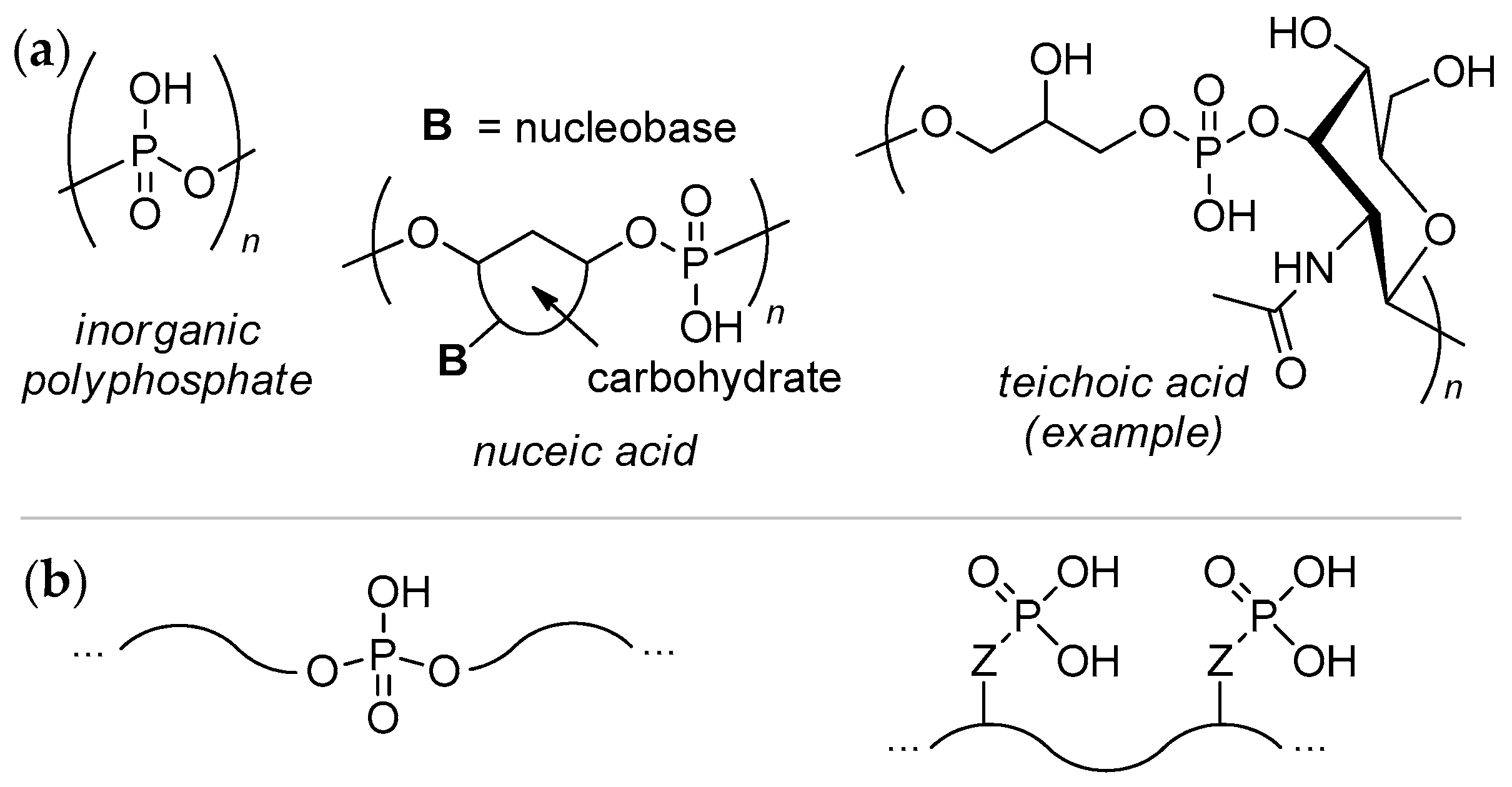
Polyacids containing –P(O)(OH)– fragment in the polymer backbone, or polyphosphodiesters (PPDEs), hold a special place among natural and synthetic polymers. The structural similarity of PPDEs to natural nucleic and teichoic acids, biocompatibility of PPDEs and their mimicking to biomolecules providing the ‘stealth effect’, high bone mineral affinity of PPDEs, and adjustable hydrolytic stability of PPDEs are the basis for various biomedical, industrial and household applications. Actual synthetic approaches to PPDEs are based on incredibly rich chemistry of organic phosphates and phosphonates, and include modern techniques such as catalytic ring-opening polymerization (ROP), acyclic diene metathesis (ADMET) polycondensation, and others.
- biocompatibility
- biodegradable polymers
- polyphosphodiesters
- ring-opening polymerization
- polycondensation
1. Introduction
2. Synthetic Approaches to Polyphosphodiesters: An Overview
In a recent review,[5] Iwasaki presented several important examples of the synthetic approaches to PCPAs. In this section, researchers have tried to enhance, refine and discuss alternative synthetic approaches to polyphosphodiesters. The synthesis of the most simple polyphosphodiesters, poly(ethylene phosphoric acid) (PEPA) and poly(1,3-propylene phosphoric acid) (1,3-PPPA), was reported by Penczek’ group back in 1976.[21] To date, multiple approaches to polyphosphodiesters have been developed. The most evident synthetic pathway is based on the interaction of phosphoric acid with diols reviewed by Penczek et al. in 2015[1] or on transesterification of dialkyl (or diaryl) phosphonates followed by oxidation of P–H bonds.[22][23][24][25][26][27][28] Ring-opening polymerization (ROP) of strained cyclic phosphonates (containing P–H bonds) and phosphates, followed by post-modification (oxidation or hydrolysis/hydrolytic thermolysis, respectively) is another efficient pathway to polyphosphodiesters.[29][30] Meanwhile, modern methods of the construction of hydrocarbon fragments of the PCPA backbone, i.e., metathesis polycondensation and polymerization,[31][32][33][34][35] should not be dismissed (Scheme 2). Note that the use of acyclic diene metathesis (ADMET) polycondensation in the synthesis of ‘precision polymers’ was the subject of review by Schulz and Wagener.[36]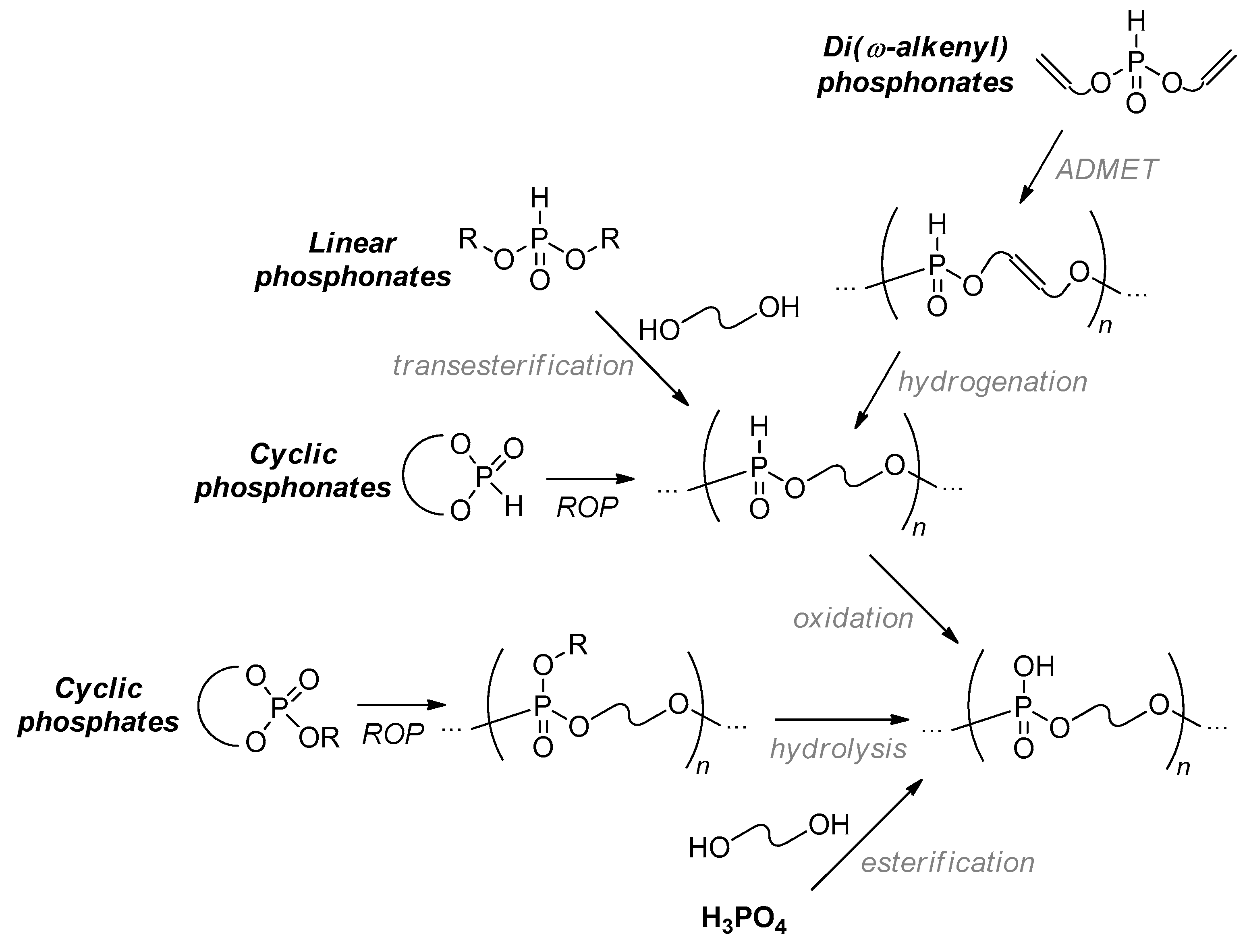
3. Polycondensation and Related Methods
3.1. Reactions of H3PO4 with Diols and Polyols
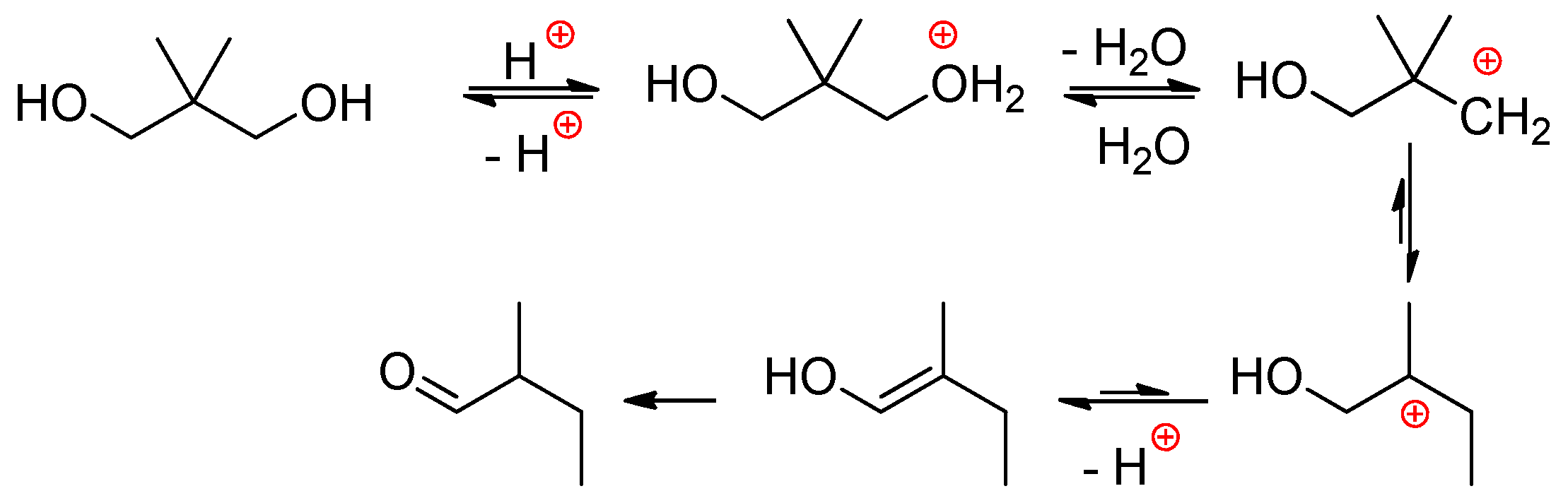
Phosphoric acid H3PO4 is a relatively weak tribasic acid (pKa1 = 2.15, pKa2 = 7.09, pKa3 = 12.32). With the transition to pyrophosphoric acid H4P2O7, one can note a substantial increase of acidity (pKa1 = 1.0, pKa2 = 2.0) and, therefore, reactivity of H4P2O7 in comparison with H3PO4. Poly(phosphoric acid) is a well-known ‘superacid’; however, its use in the synthesis of PCPAs is essentially restricted by the requirements of the hydrolytic stability of PCPAs that implies the absence of di-/oligophosphate fragments in the main polymer chain. In this way, successful synthesis of PPDEs was limited by the use of H3PO4 and H4P2O7 in polycondensation with diols and polyols. This approach was developed mainly by Penczek and coll who studied direct condensation of H3PO4 with ethylene glycol.[37][38][39] The following steps were detected during this reaction:-
The reaction starts by the relatively slow dimerization of H3PO4 with a formation of H4P2O7 (and higher polyphosphoric acids) at 100 °C within 40 h, during this stage the water was removed either in the stream of neutral gas or azeotropically with heptane.
-
After the addition of EG at 100 °C, H4P2O7 transformed to H3PO4 immediately, and the first phosphorylation reaction within additional 80 h was the formation of HOCH2CH2OP(O)(OH)2 and (HOCH2CH2O)2P(O)OH, triesters were formed in minimal amounts.
-
Activation of the monophosphate esters (end groups) at any polymerization degree with H3PO4 proceeds via conversion of monoesters into pyrophosphoric acid esters –OCH2CH2OP(O)(OH)–OP(O)(OH)2 that represent reactive acidic sites.
-
The polycondensation product is mostly linear with a structure of PEPA –(OCH2CH2OP(O)(OH))n–.
-
Some branch points (triesters) are formed only at high temperature and prolonged polycondensation time.
3.2. The Reaction of Dichlorophosphates with Diols
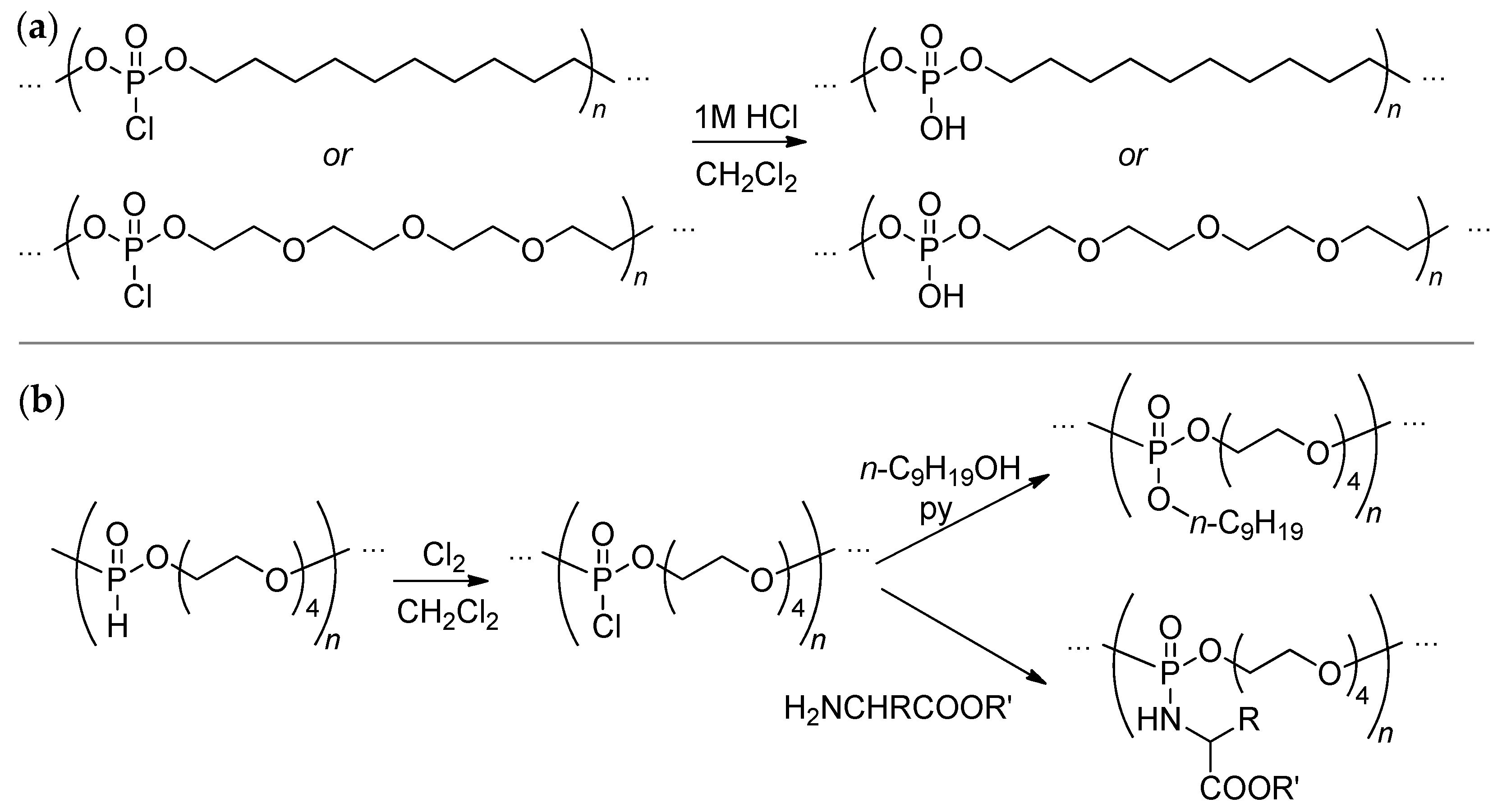
Glycolysis of PET with a formation of bis(2-hydroxyethyl)phthalate is the most efficient method of chemical recycling of this polymer.[46][47] The reaction of bis(2-hydroxyethyl)phthalate with Cl2P(O)OR (R = Me, Et) resulted in the formation of copolymers, further treatment by terephthaloyl chloride and NaI/acetone allowed for a copolymer containing >P(O)–OH fragments to be obtained (Scheme 4).[48] However, the current trends in developing actual synthetic approaches to biodegradable materials imply the abandonment of chlorine-containing reagents, and therefore dichlorophosphates are not currently used in the synthesis of polyphosphodiesters.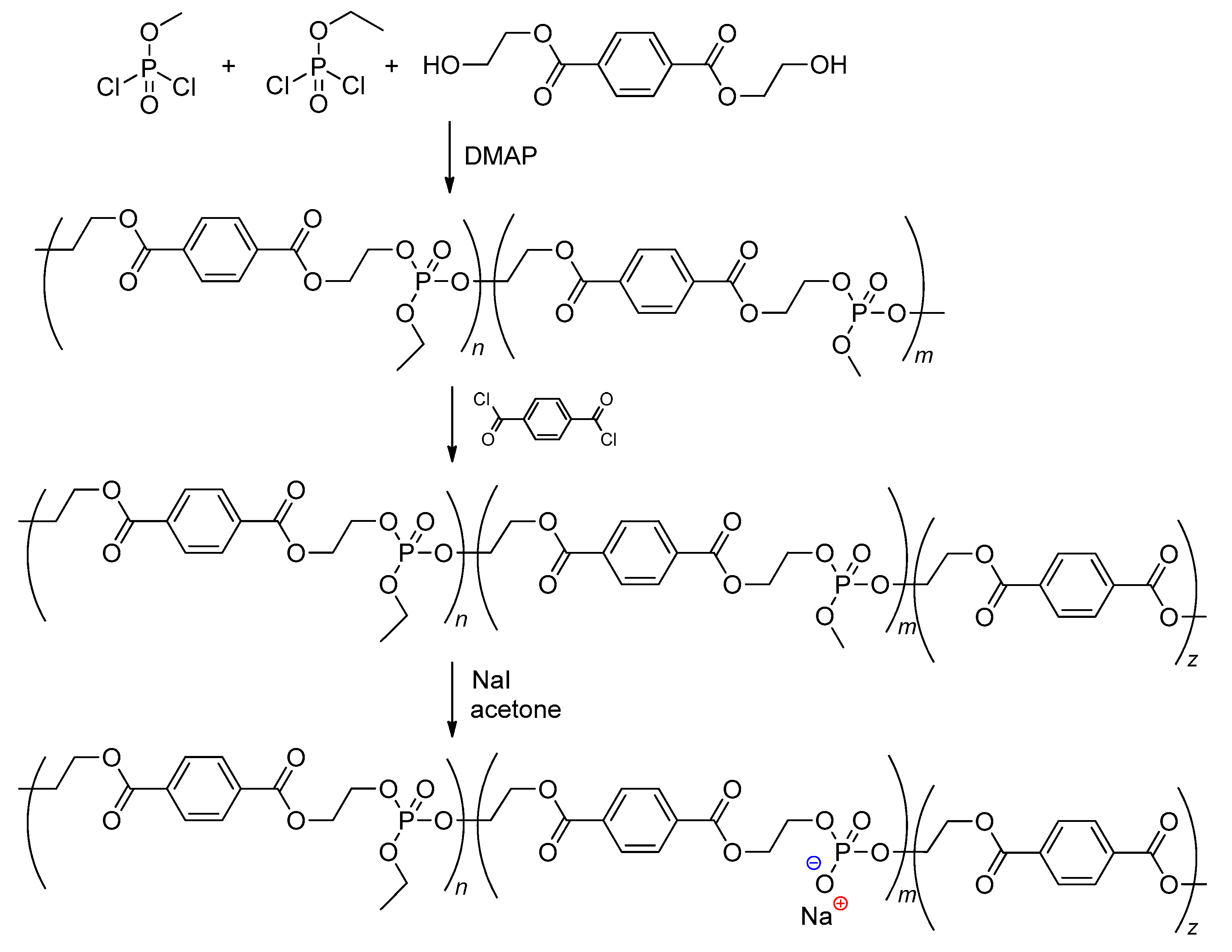
3.3. Reaction of Dialkyl (or Diaryl) Phosphonates with Diols and Post-Modification
Since polymers with –O–P(O)H–O– fragments can be easily and almost quantitatively oxidized to corresponding poly(phosphodiesters) containing –O–P(O)(OH)–O– fragments,[21][22][49] polycondensation of dialkyl phosphonates (RO)2P(O)H with diols can be considered as a prospective method of the synthesis of polyphosphodiesters. However, when using propane-1,3-diol, a six-membered cyclic phosphonate is formed at elevated temperatures, and further low-temperature ROP is needed for the synthesis of PPDE.[50] In addition, Penczek and coll have proposed that for the successful synthesis of high-MW polymer the alcohol ROH has to be removed as fast as possible.[51] Relatively high-MW poly(alkylene phosphonates) (Mn = 9.3–28 kDa) were obtained by the reaction of (MeO)2P(O)H with HO–(CH2)n–OH (n = 5–10, 12).[22] Polytransesterification of dimethyl phosphonate (MeO)2P(O)H and poly(ethylene glycol)s with Mn 200 Da (PEG200) and 600 Da (PEG600) resulted in copolymers with Mn = 3.5 and 7.1 kDa, respectively;[23][24] similar results were obtained using PEG400, transesterification was conducted within 5 h at 135 °C under atmospheric pressure, and then 4 h at 160 °C plus an additional 15 min at 185 °C in vacuo (1 Torr), degree of polymerization (DPn) was 28.[25] The reaction of H(OCH2CH2)13O H with (MeO)2P(O)H also resulted in the formation of the polymer (Mn = 13.5 kDa).[26] Poly(1,2-propylene glycol) (PPG)-based oligo(alkylene phosphonate)s with DPn 12, 6 and 5 were synthesized with the use of PPG400, PPG1200 and PPG2000, respectively.[27] Triblock copolymers mPEG750-b-[(P(O)(H)O(CH2)6]17-b-mPEG750 and mPEG2000-b-[(P(O)(H)O(CH2)6]17-b-mPEG2000 were obtained by polycondensation of (MeO)2P(O)H with HO–(CH2)6–OH (4 h at 80 °C and then 9 h at 140 °C/1 Torr, 0.05 mol% Na to form the catalyst), followed by the reaction with mPEG (140 °C/1 Torr).[52] To achieve high molecular weights of the polycondensation products, Penczek and coll proposed the use of diphenyl phosphonate in reaction with diols.[53] The reaction was conducted at 140 °C with the elimination of the phenol, and PPDEs with Mn up to 40 kDa were obtained (Scheme 5).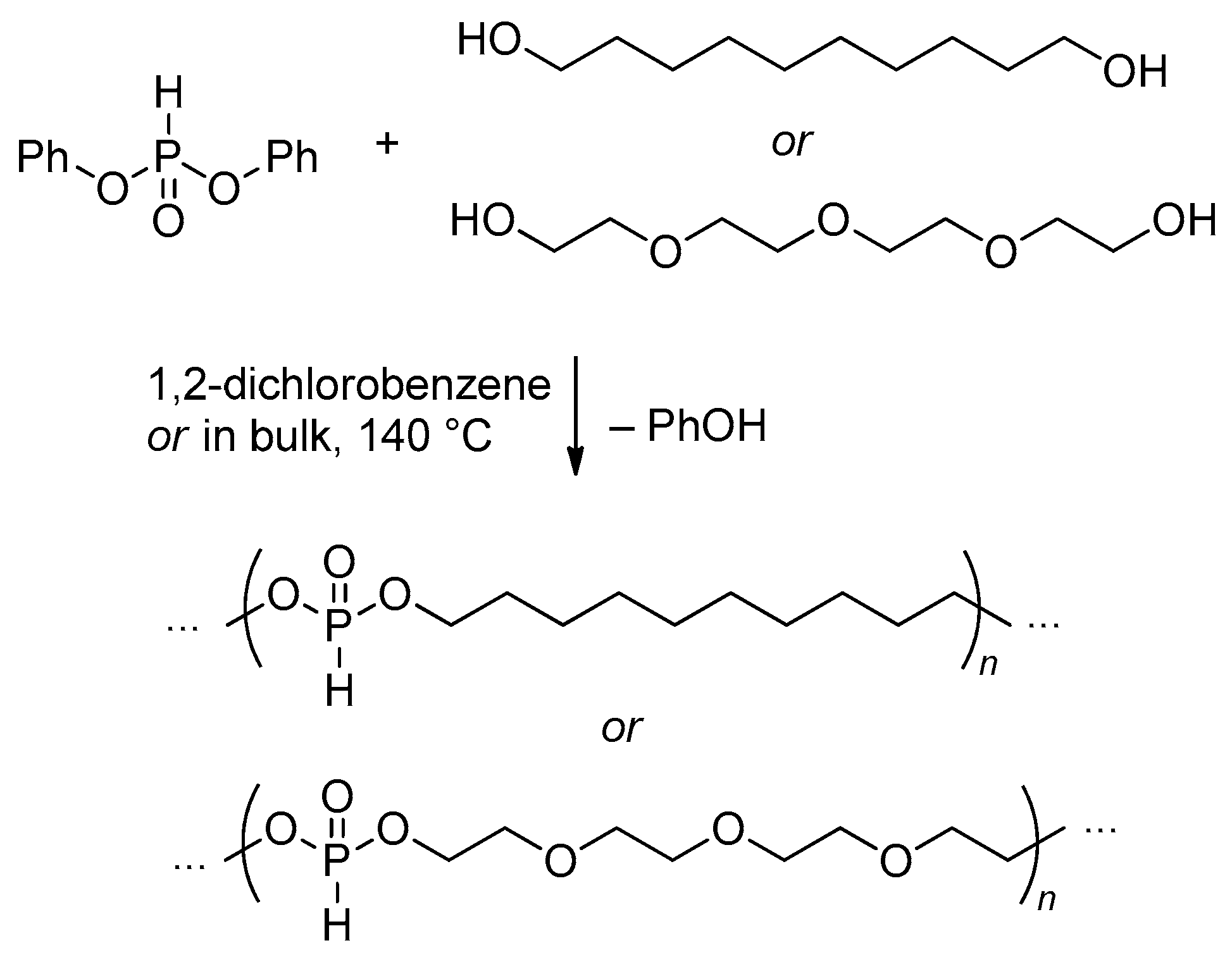
3.4. Polycondensation of (ω-Hydroxyalkyl)phosphonic Acids
In 2020,[58] Penczek and coll. have shown that hydroxymethyl phosphonic acid can act as a catalyst and initiator of the ROP of ε-caprolactone (εCL) with the formation of εCL oligomers containing reactive groups on both ends of the macromolecule. Very recently they demonstrated that these oligomers can be subjected to polycondensation at 100–110 °C with a formation of PPDEs (Mn up to 25 kDa) (Scheme 8) with mostly linear microstructure (31P NMR data).[59]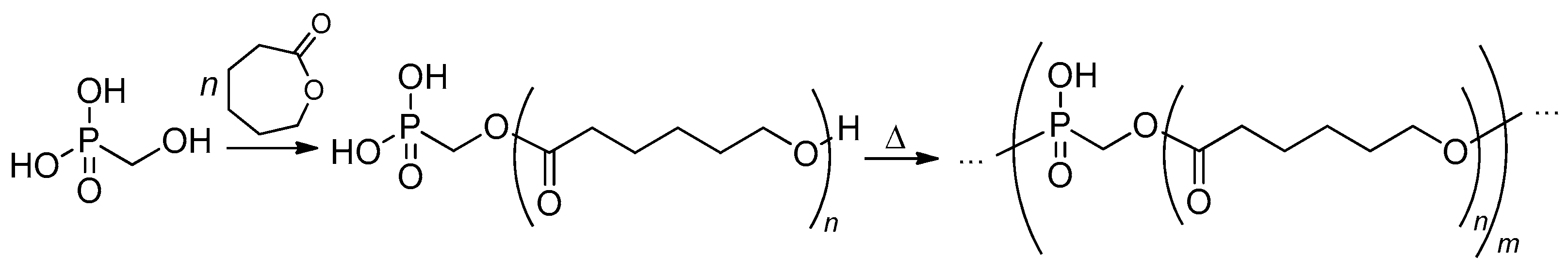
4. ROP of Cyclic Phosphorus-Containing Monomers and Post-Modification
4.1. Synthesis of Cyclic Phosphorus-Containing Monomers
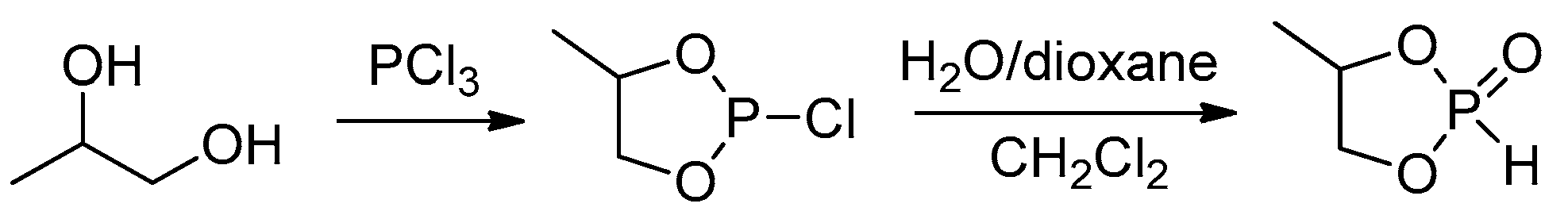
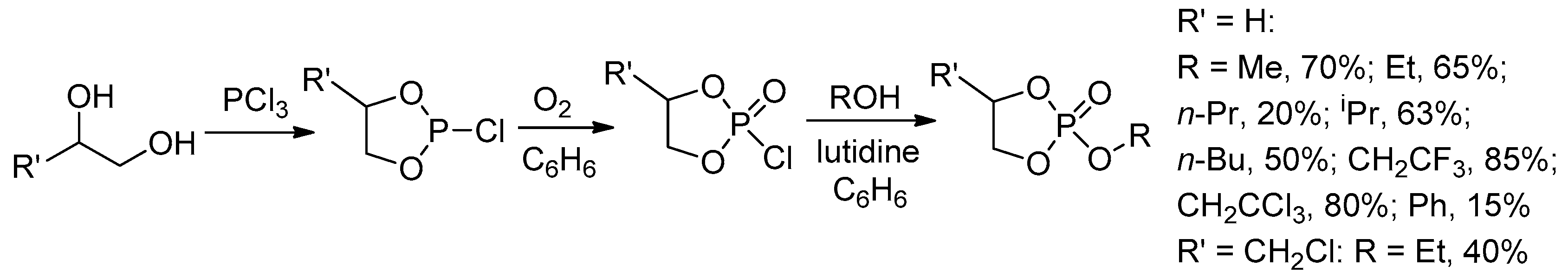
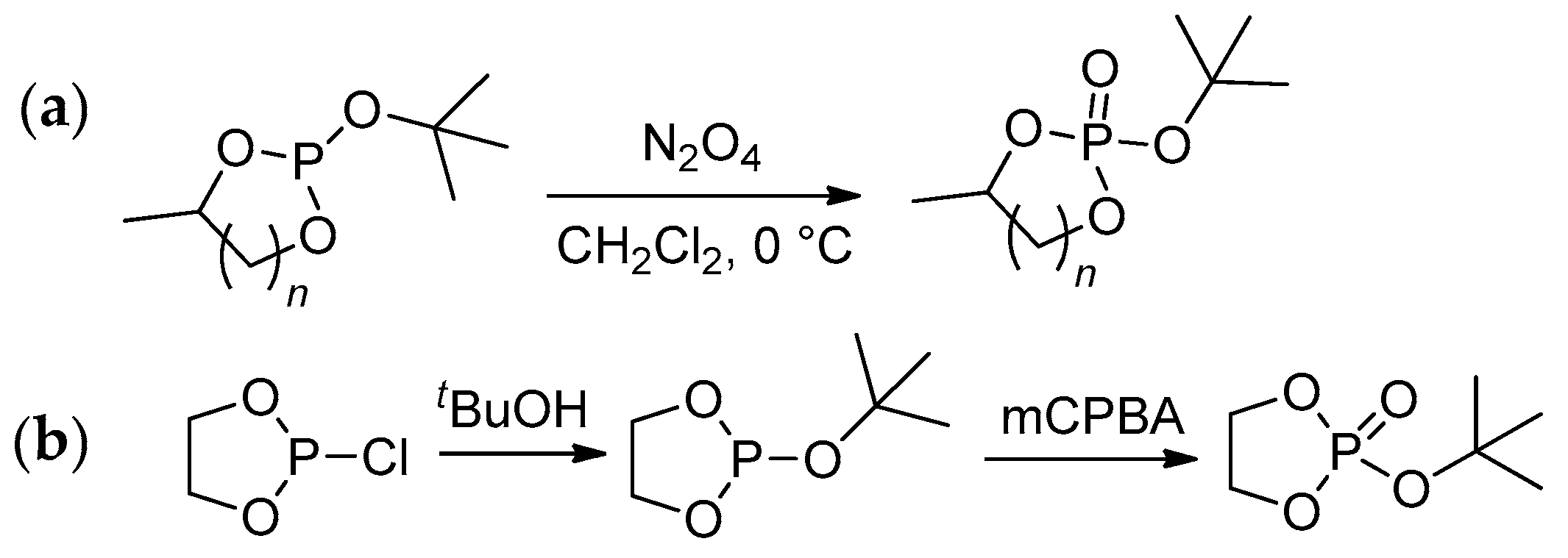
The key stage of the preparation of both cyclic phosphonates and cyclic phosphates is a reaction of diols with PCl3 resulting in cyclic chlorophosphites[60] that can be hydrolyzed with the formation of cyclic phosphonates (Scheme 9a) or oxidized to chlorophosphates with subsequent substitution of Cl atom by alkoxy fragment that results in cyclic phosphates (Scheme 9b). In some cases, the synthesis of cyclic phosphates is based on reverse reaction sequence, i.e., substitution of Cl in chlorophosphite followed by oxidation (Scheme 9c).[61] Cyclic phosphonates can also be synthesized by the reaction of diols with dialkyl phosphonates (Scheme 9d).[62][63]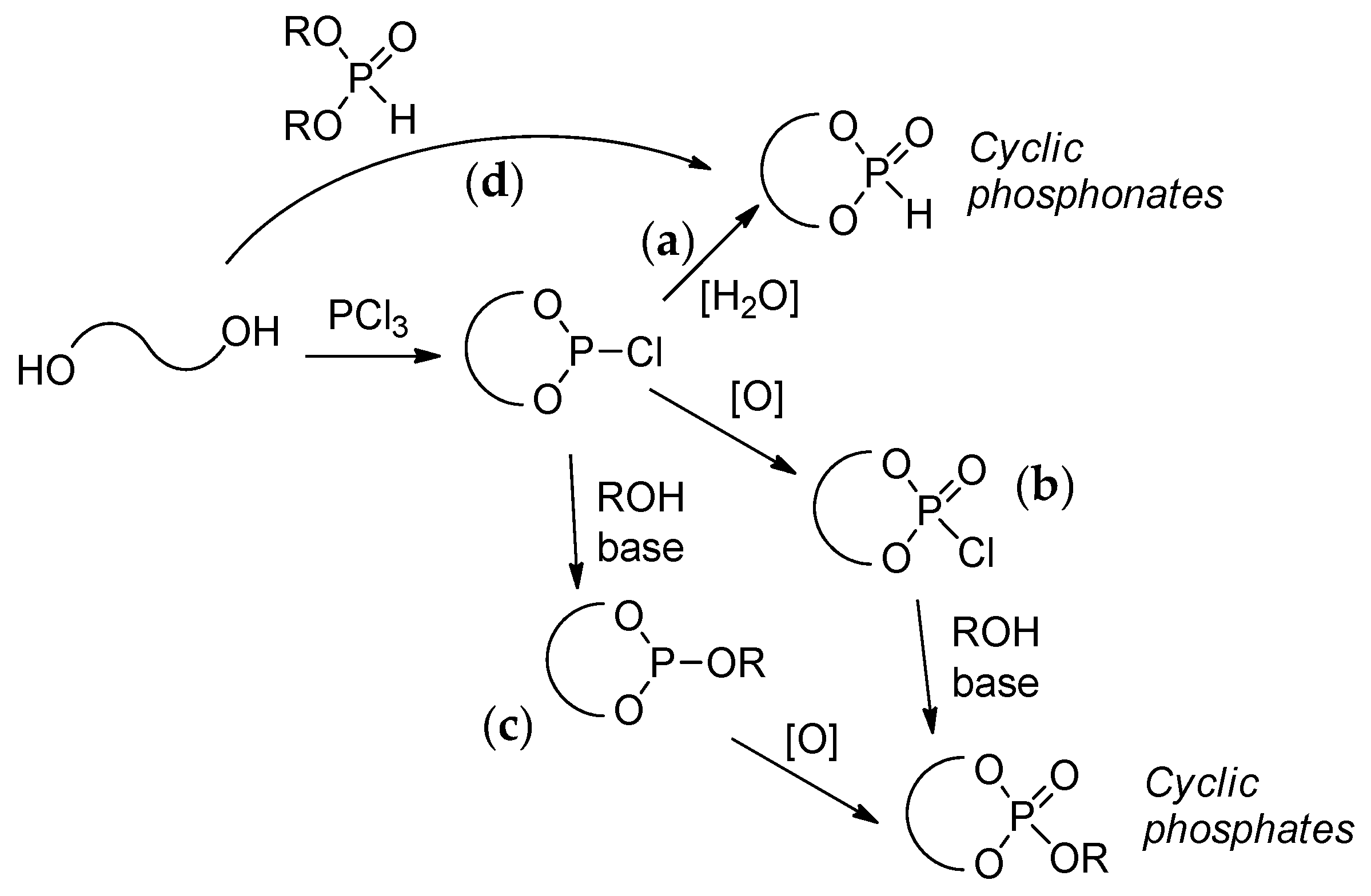
4.2. ROP of Cyclic Phosphorus-Containing Monomers
ROP of cyclic phosphonates and phosphates (Scheme 13a) represents the common strategy of the controlled synthesis of functional biodegradable polymers.[29][30] This process is subject to the general thermodynamic rules for the ROP of cyclic monomers[72] that predict high reactivity of more strained five-membered cycles[66][73][74][75] and temperature-dependent reactivity of six-membered cycles.[74][75] Different catalysts have been used successfully in controlled ROP of cyclic phosphonates and phosphates with the formation of polyphosphoesters (PPEs) (Scheme 13b). The data on the synthesis of polymers suitable for post-modification to polyphosphodiesters are summarized in Table 1.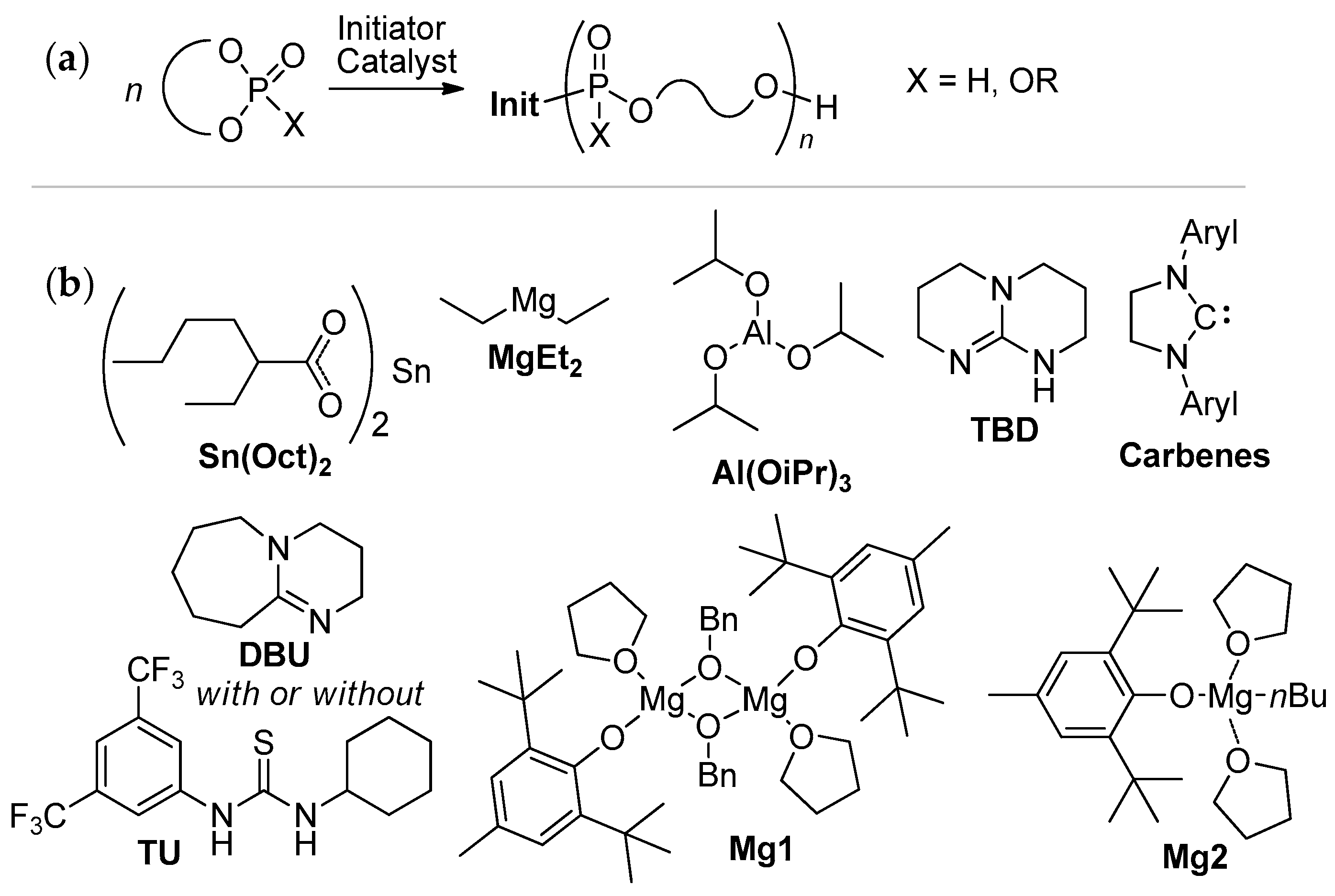
| Entry | Monomer | Catalyst | Reaction Conditions/Conversion, % | Mn, kDa | DPn a | ÐM | Refs. |
|---|---|---|---|---|---|---|---|
| 1 | 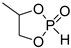 |
iBu3Al | CH2Cl2, 20 °C | - | - | - | [64] |
| 2 | 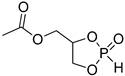 |
iBu3Al | CH2Cl2, –20 °C, 6 h/80 | [49] | |||
| 3 | 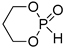 |
iBu3Al | CH2Cl2, from 0 to 20 °C | 90 | 740 | - | [21][75] |
| 4 | 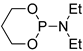 |
tBuOK | THF, 20 °C, several days/99 | - | - | - | [76] |
| 5 | 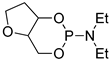 |
tBuOK | C6H6, 20 °C, several days/99 | - | - | - | [71] |
| 6 | 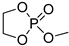 |
iBu3Al | CH2Cl2, from –20 to 20 °C | 30–100 | - | - | [21] |
| Et2Mg | CH2Cl2, from –20 to 20 °C | 30–100 | - | - | [21][66] | ||
| DBU/TU | CH2Cl2, 20 °C, 15 min/83 | 9.2 | 68 | 1.17 | [61] | ||
| DBU/TU | CH2Cl2, 0 °C, 1.4 h/92 | - | 97 | - | [77] | ||
| Mg1 | CH2Cl2, –20 °C, 5 min/99 | 9.5 | 70 | 1.35 | [61] | ||
| TBD/BnOH | CH2Cl2, –20 °C, 5 min/99 | 9.3 | 68 | 1.24 | [61] | ||
| TBD/BnOH | CH2Cl2, 1 eqiv. TMP, –20 °C, 5 min/99 | 6.4 | 47 | 1.13 | [78] | ||
| DBU/Cholesterol | CH2Cl2, 20 °C, 5 h/ | [79][80] | |||||
| 7 | 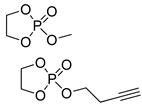 |
DBU/EtOH DBU/MeOH |
9:1 comonomer ratio, CH2Cl2/– - |
- - |
38, 85, 127 73 |
- - |
[81] [82] |
| 8 | 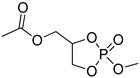 |
iBu3Al | CH2Cl2, 0 °C | 25 | 119 | - | [49] |
| 9 | 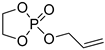 |
DBU/TU BnOH | toluene, 0 °C, 10 min/80 | - | - | - | [83] |
| DBU/TU mPEG5000 | toluene, 0 °C, 10 min/80 | 7.5 | 16 | <1.2 | [84] | ||
| 10 | 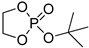 |
Et2Mg | C6H6, 40 °C, 10 h/80 | 25 | 139 | - | [70] |
| Mg1 | CH2Cl2, 20 °C, 18 h | 6.4 | 36 | 1.19 | [61] | ||
| Mg1 | CH2Cl2, 20 °C, 18 h | – | 63 | – | [85] | ||
| Mg2/mPEG5000 | CH2Cl2, 20 °C, 30 h | 3.6 | 13 | 1.45 | [86] | ||
| CH2Cl2, 20 °C, 30 h | 8.1 | 49 | 1.48 | [86] | |||
| 11 | 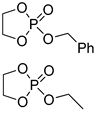 |
iBu3Al | 1:10 comonomer ratio, bulk/69.3 | 6.0–7.0 | - | - | [69] |
| TBD/BnOH TBD/ Cholesterol |
5:95–20:80 comonomer ratio, toluene 4:96 and 17:83 comonomer ratios, CH2Cl2 |
9.5–11.9 4.6; 6.4 |
- - |
1.45–1.62 1.3; 1.2 |
[87] [88] |
||
| 12 | 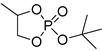 |
Et2Mg | C6H6, 40 °C, 10 h/90 | 25 | 139 | n.d. | [70] |
| 13 | 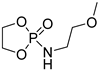 |
TBD/BnOH | CH2Cl2, 0 °C, 1 min/99 | 13 | 72 | 1.17 | [89] |
| 14 | 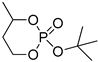 |
Et3Al/H2O | C6H6, 40 °C/50 | - | - | - | [70] |
4.3. Post-Modification of the Poly(alkylene phosphonate)s
Oxidation of the polymers containing –P(O)H– fragments in the main chain represents a promising synthetic approach to PCPAs. In earlier studies, N2O4 in CH2Cl2 was found to be an efficient oxidizing reagent (Scheme 14), the resulting polyacids precipitated.[21][49][64][75] Wang et al. reported the use of DMF as a solvent for oxidation.[94] It is worth pointing out here that the formation of HNO3 during oxidation may assist the cross-linking between PCPAs’ polymer chains thereby decreasing the control on polymer MWD and architecture, thus, for example poly(1,2-propylene phosphoric acid) (1,2-PPPA) synthesized in DMF had Mw = 12.9 kDa and Ð = 2.6.[94] In an early work of Penczek’s group, the reaction with O3 was proposed as an efficient method of the transformation of poly(alkylene phosphonate)s to corresponding polyphosphates (Scheme 15).[71] Note that starting poly(alkylene phosphonate) was obtained via ROP of cyclic phosphoramidite followed by acid hydrolysis of the polymer obtained.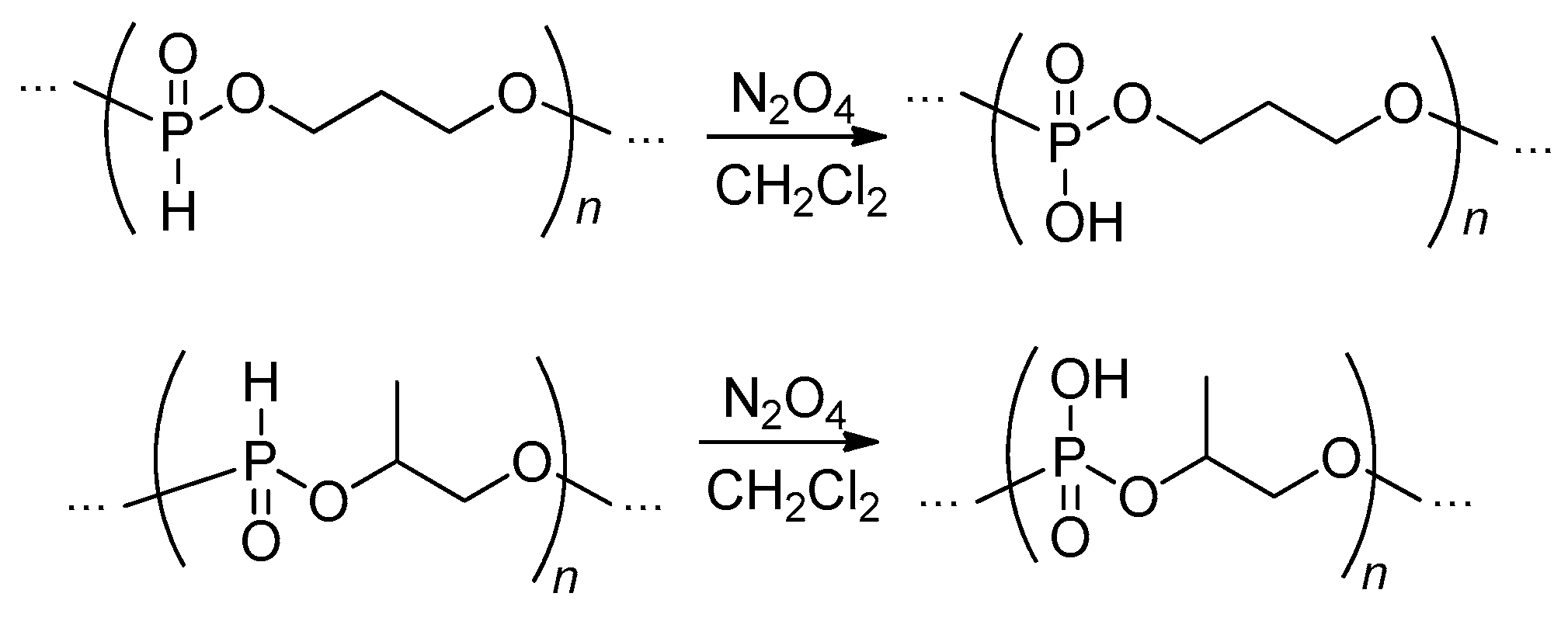
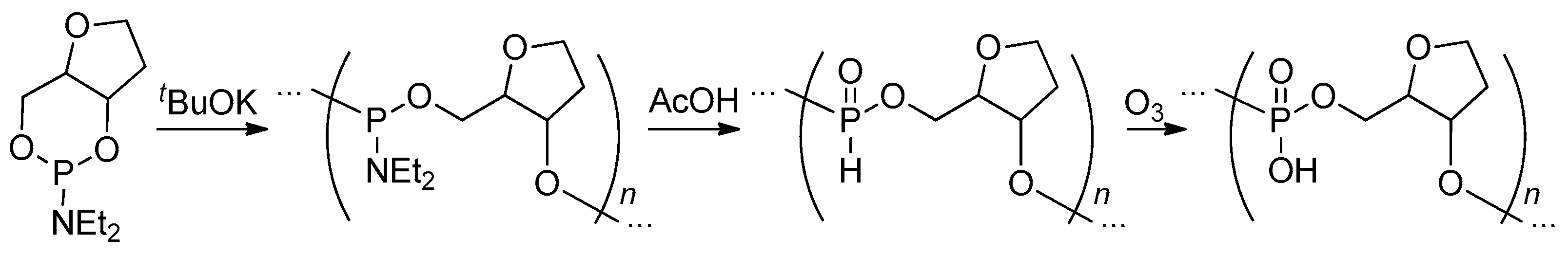
4.4. Post-Modification of Poly(alkylene phosphate)s
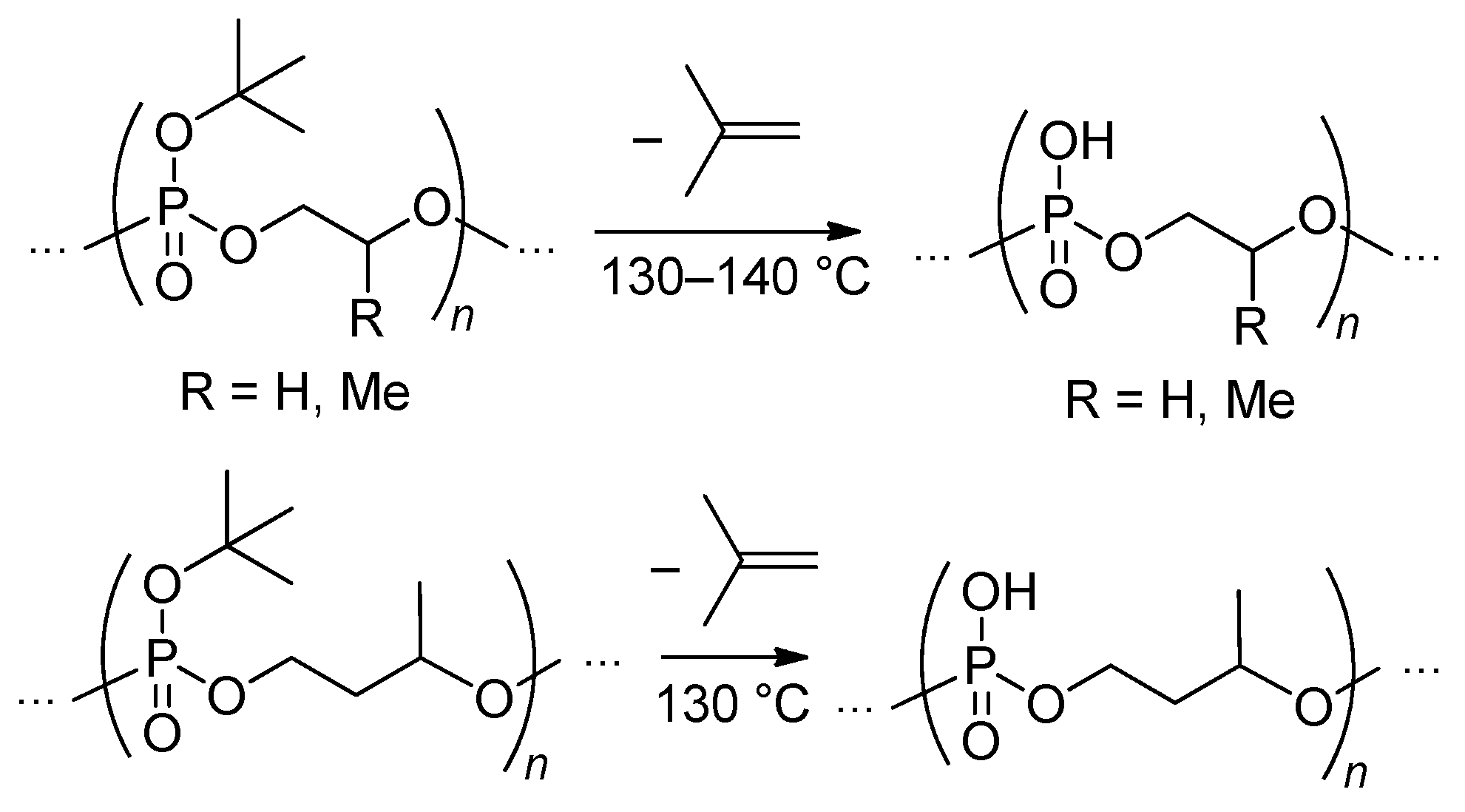
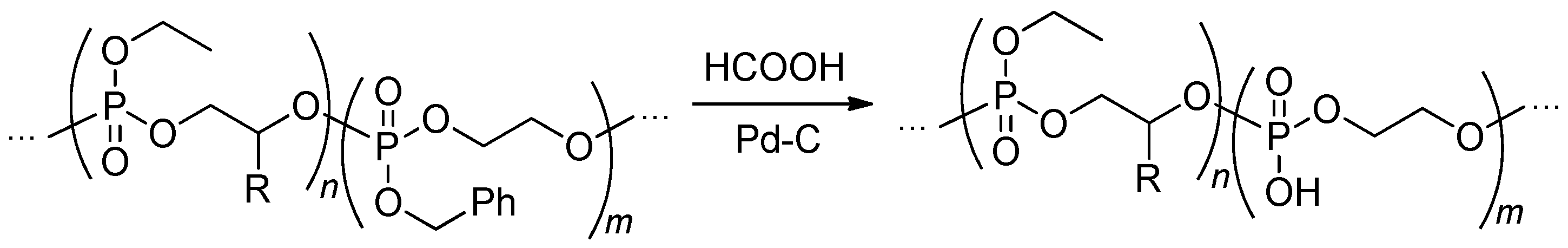
The most evident synthetic pathway to PEPA is based on hydrolysis of the ester side groups with a maintaining of poly(alkylene phosphate) backbone (Scheme 16). The first attempt of such hydrolysis was made by Gehrmann and Vogt back in 1981 with the use of 1-oxo-2,6,7-trioxa-1-phosphabicyclo [2.2.l] heptane homopolymer of unidentified structure.[95]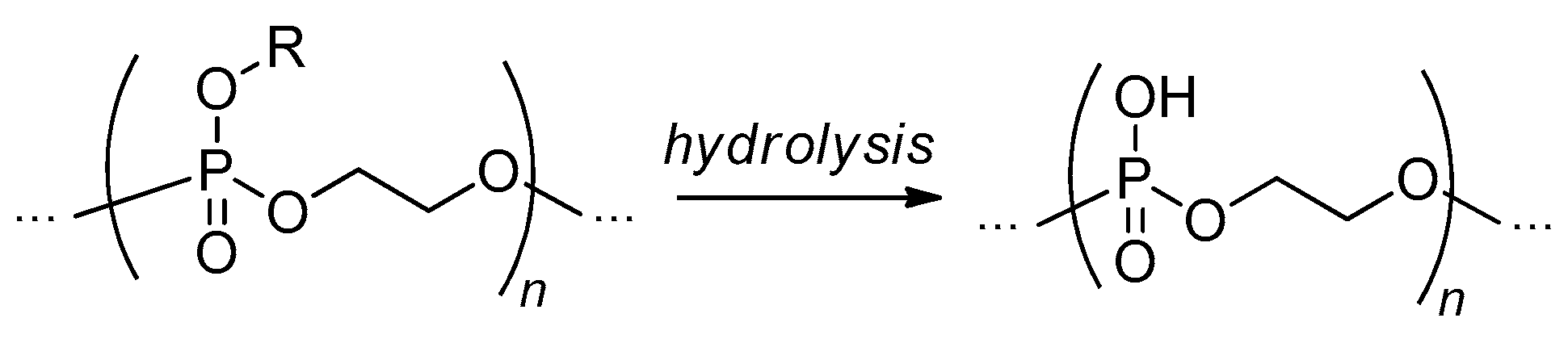
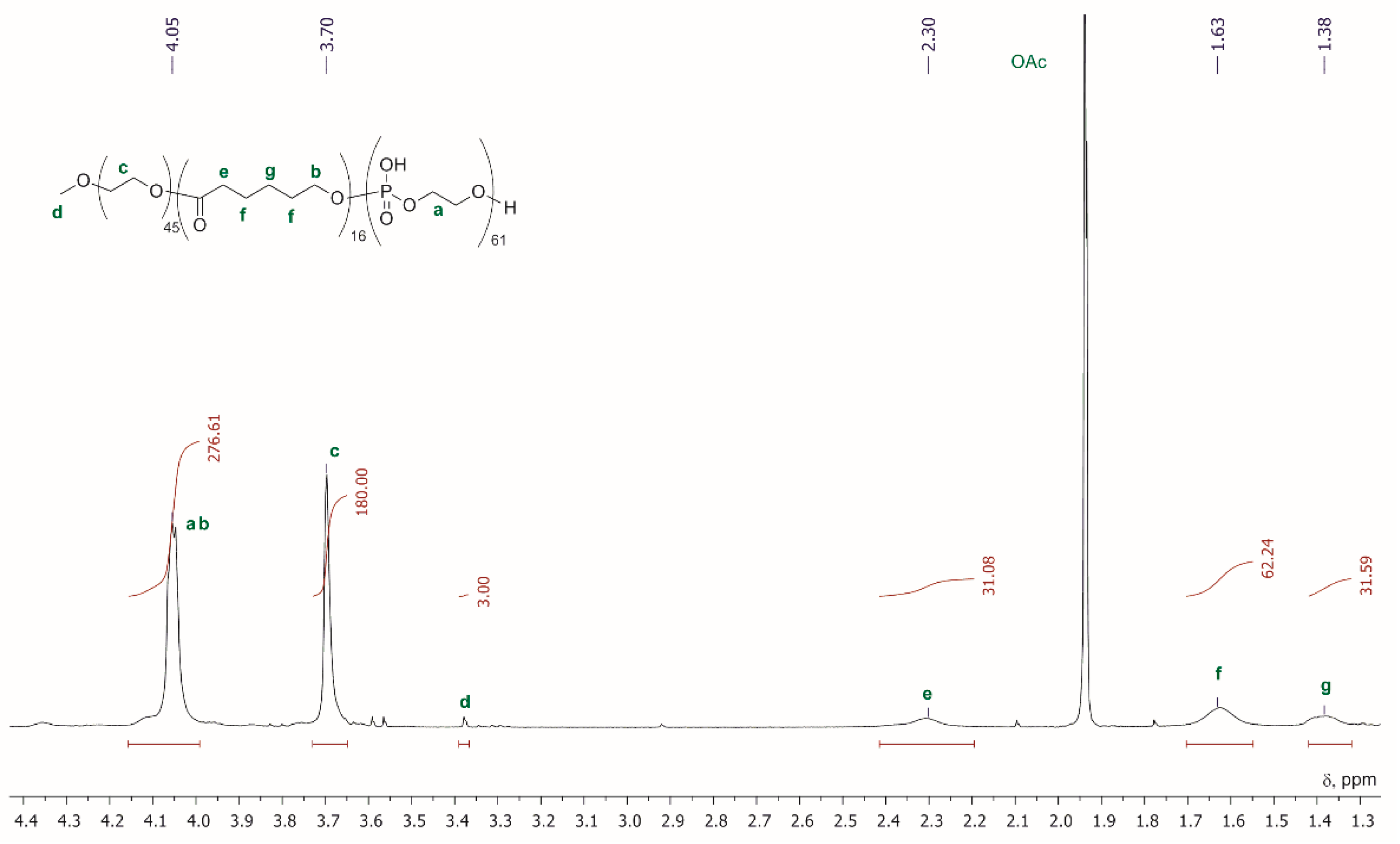
-
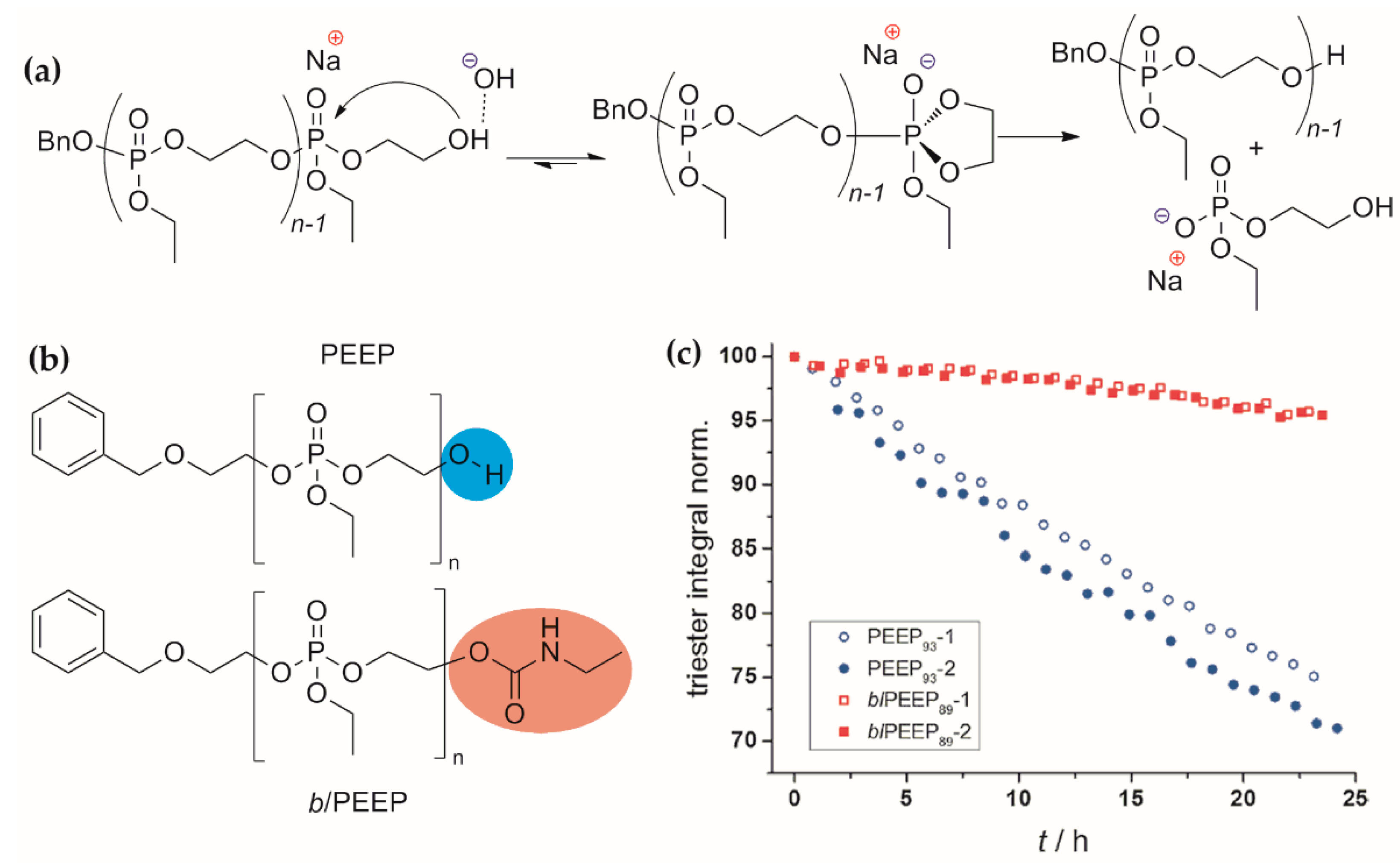
Loss of control over polymer architecture and MWD: sterically non-hindered cyclic phosphates can form highly branched poly(alkylene phosphate)s. Switching between the ‘living’ (linear polymer, ĐM~1) and ‘immortal’ (transesterification of the polymer chain, branched polymer, ĐM > 1) ROP modes can occur at elevated temperatures and/or in case of wrong catalyst’ choice. Moreover, even in the presence of ‘good’ catalysts, complete conversion of the monomer greatly increases the risk of subsequent transesterification.
-
This is why better chain control can be achieved when using sterically hindered cyclic phosphates, e.g., tBuOEP, despite its minor synthetic accessibility and very low reactivity that limits the use of this monomer in the synthesis of stat- and block-copolymers.
-
The use of cyclic phosphonates eliminates the problem of branching and DPn control, but severe oxidation of the P–H bond at the final stage puts the end to a convenient option to introduce biomolecules or usable functional groups at the stages of ROP initiation or termination.
-
The nature of the catalytic ROP imposes severe restrictions on the nature of the side substituent R in the molecule of cyclic phosphate (Scheme 16). So, for example, the –CH2CH2CN group, widely used in automated (!) synthesis of DNA analogs[102] and in synthesis of PCPAs with the use of ring-opening metathesis polymerization (ROMP),[103] has not found application in the ROP/deprotection approach to PCPAs, despite the fact that the synthesis of six-membered cyclic phosphate with this substituent was synthesized by Lapienis and Penczek back in 1977.[65]
-
Additionally, in general, between fundamental studies of the ROP/deprotection approach to PCPAs in the late 1970s–1980s (conducted for the most part by the Penczek’ group) and relatively recent works (scientific groups of Wooley, Wurm, Iwasaki, Nifant’ev), a two-decades gap in investigations is clearly visible, which affected the progress in this scientific direction.
5. Metathesis Polycondensation
In 2014 Wurm and coll. proposed an efficient synthetic approach to polyphosphodiesters based on ADMET polycondensation of bis(alkenyl) chlorophosphates, catalyzed by the first generation Grubbs catalyst.[31] In bulk polymerization, DPn of 39 was achieved, and when using 1-chloronaphthalene as a solvent, DPn was 47 and 126 for ‘chloro monomers’ containing –(CH2)2– and –(CH2)9– spacers between vinyl and phosphate fragments, respectively (Scheme 19). Functionalized PCPAs were then obtained by the reactions of poly(alkylidene chlorophosphate)s with PhOK or (2-hydroxyethyl)methacrylate (HEMA) in the presence of water.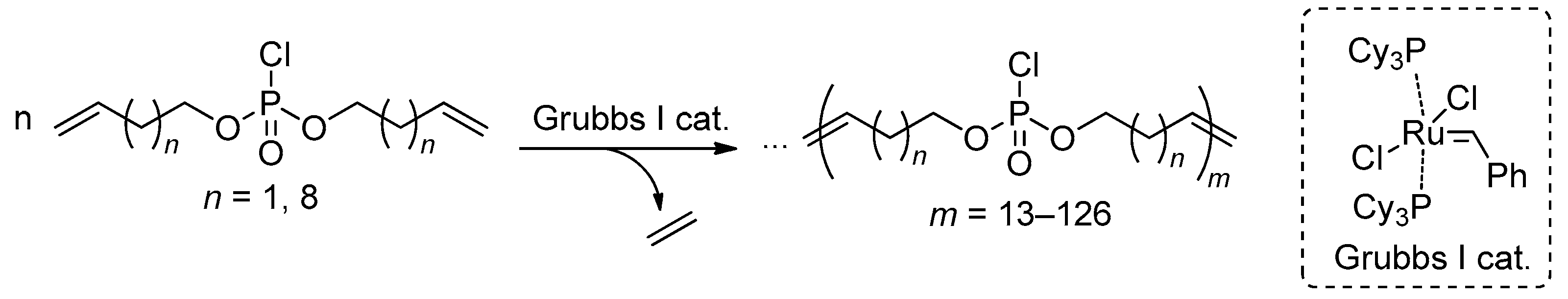
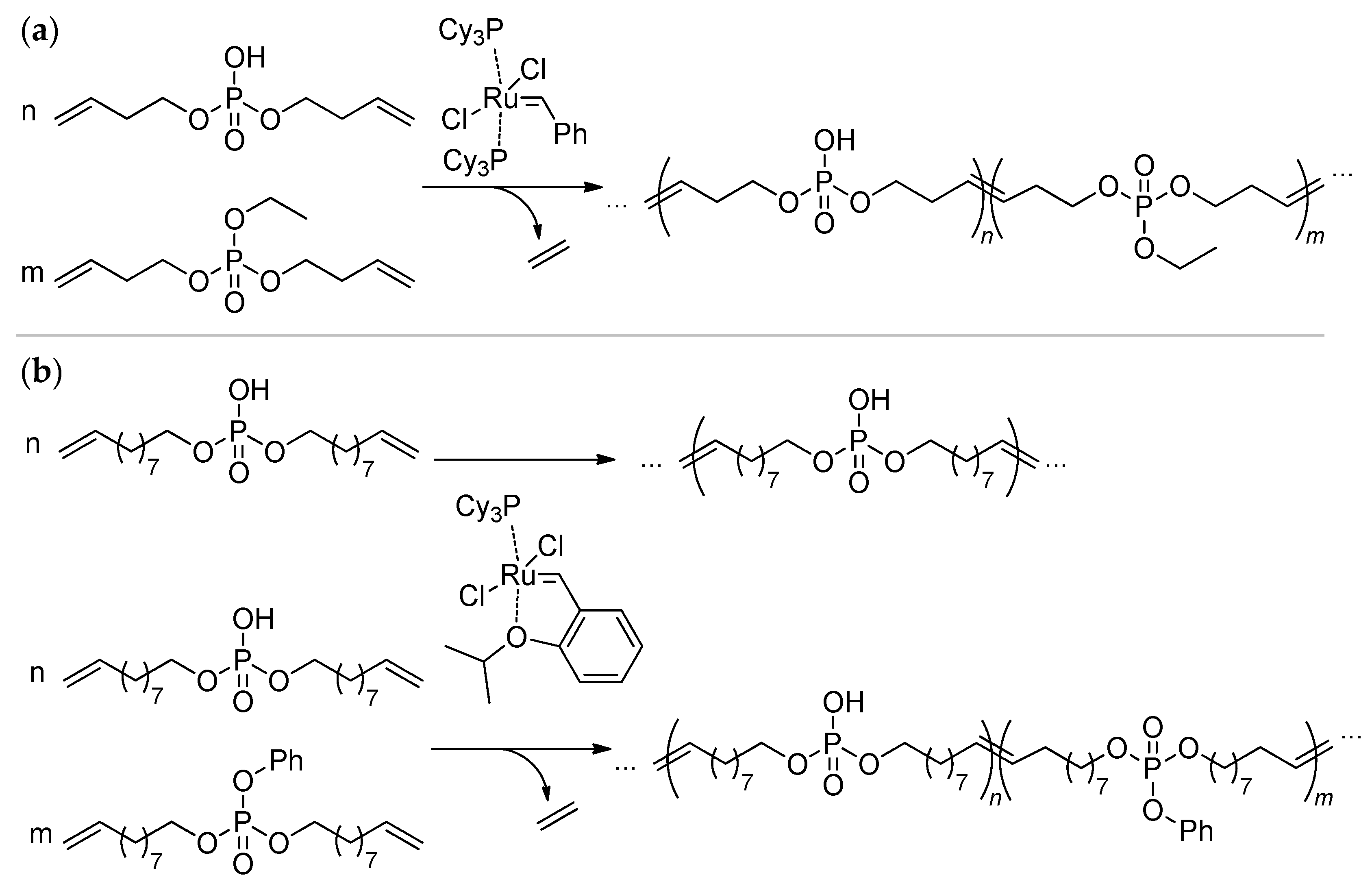
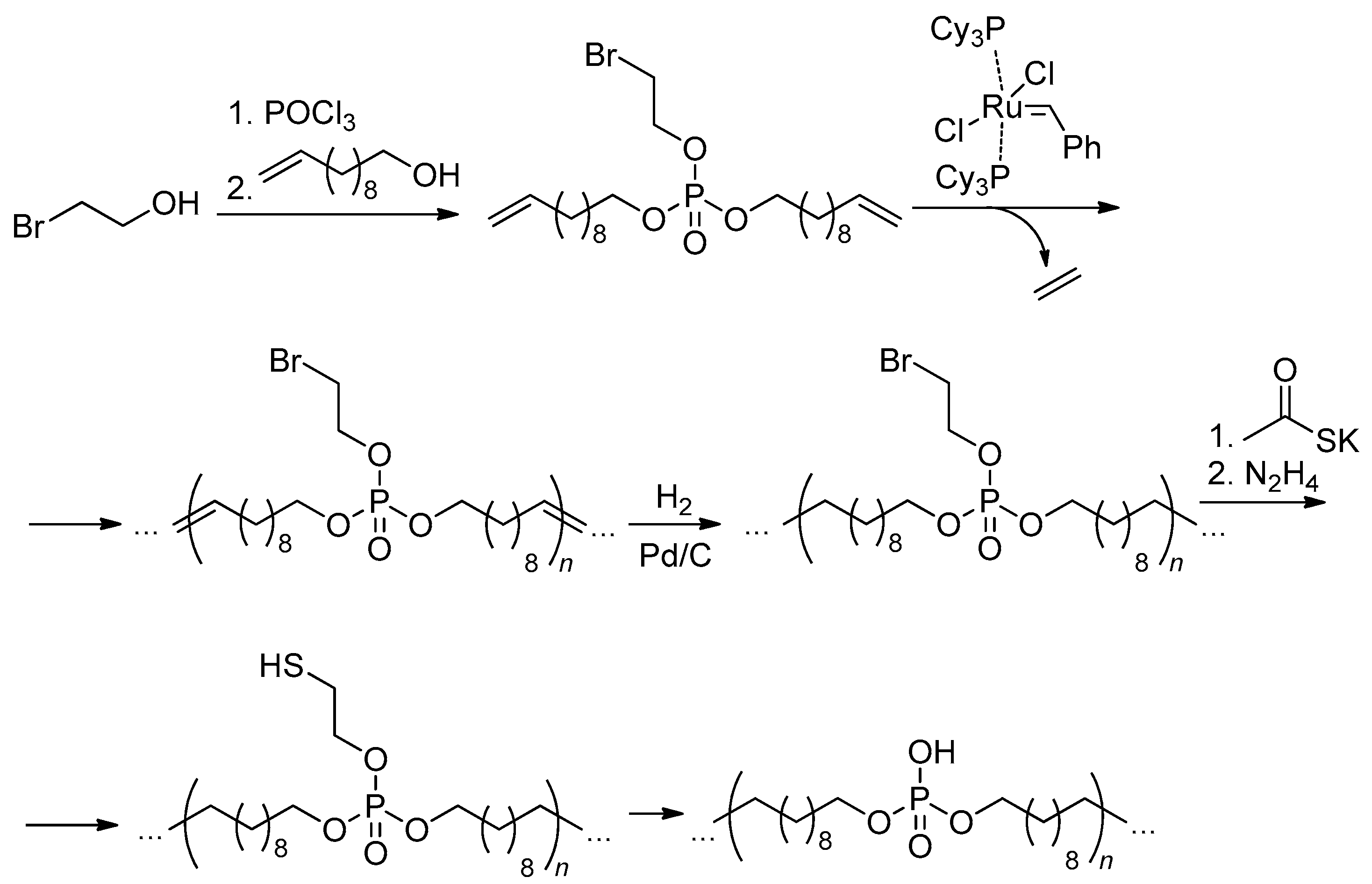
6. Other Synthetic Approaches to Polyphosphodiesters
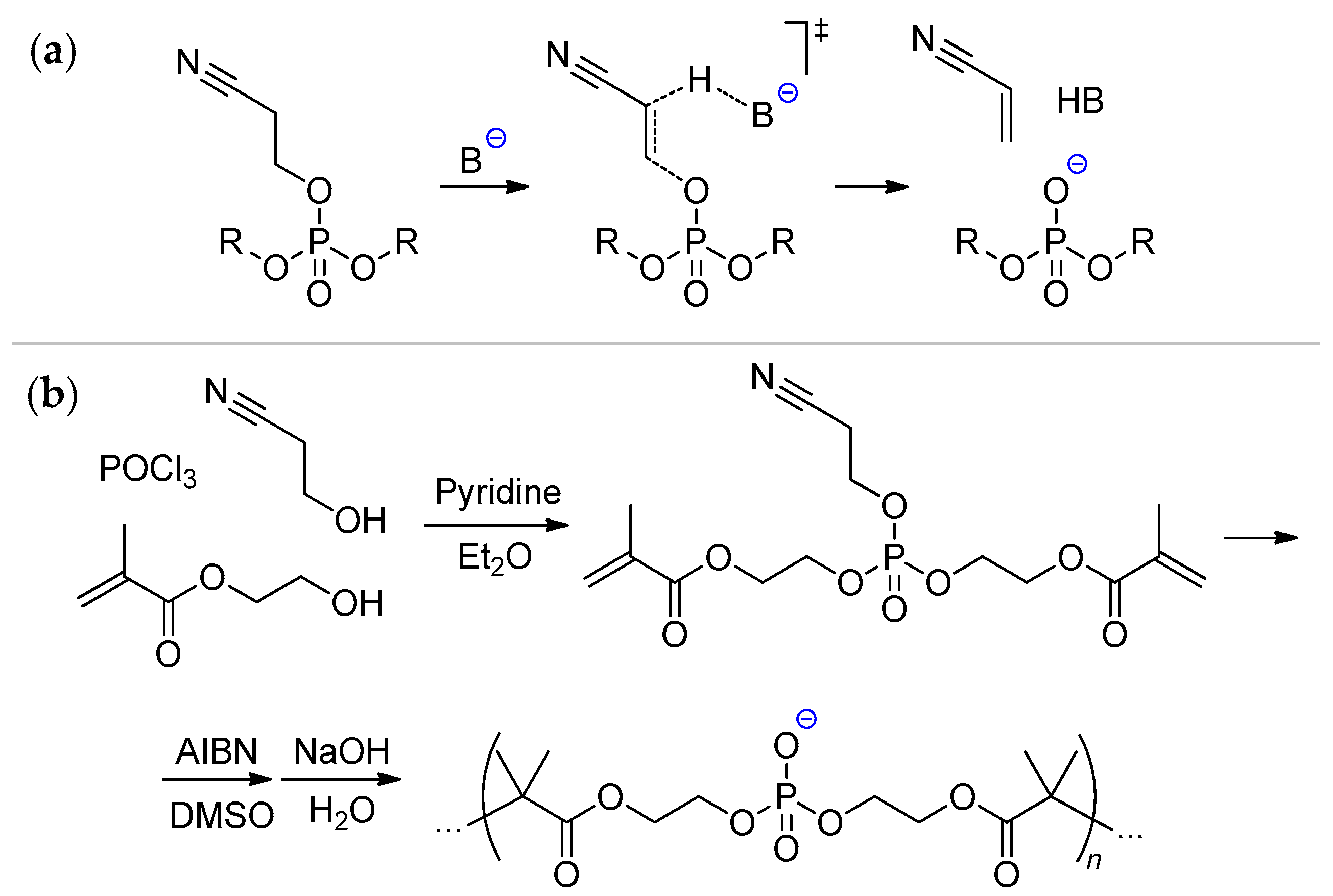
6.1. The Use of Unsaturated 2-Cyanoethyl Phosphates
6.2. Bis(methacrylate) Phosphonates and Their Post-Modification
Diliën and coll. proposed efficient synthetic approach to monomers for the synthesis of PPDE-containing hydrogels, based on the reaction of (PhO)2O(O)H or H3PO3 with 2-hydroxyethylmethacrylate (HEMA), followed by the Atherton–Todd reaction with N-tert-butyl-4-hydroxybutanamide and CCl3Br/NEt3 (Scheme 23). Free-radical polymerization of this monomer followed by thermal deprotection via elimination of stable five-membered iminoester resulted in formation of the polymers containing main-chain –P(O)OH– fragments.[104]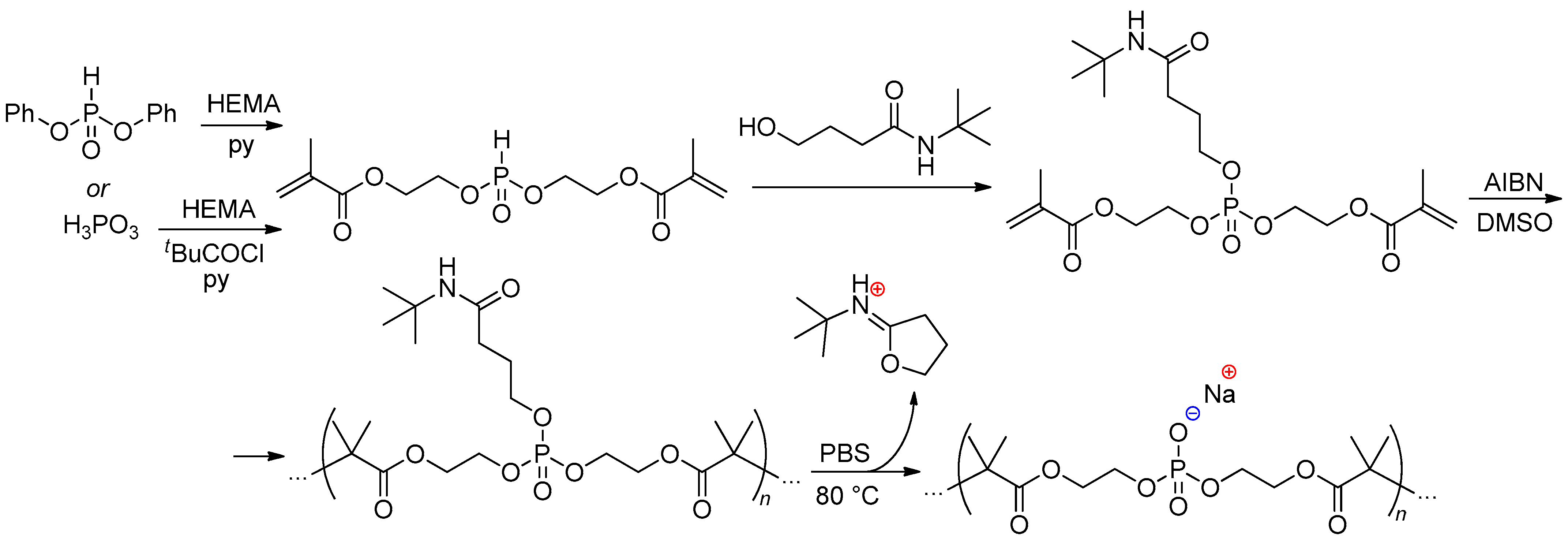
6.3. Hydrolytic Polymerization of Spiro(acylpentaoxy)phosphoranes
Saegusa and coll. have demonstrated that spiro-phosporanes can react with water to form polymers containing phosphodiester and phosphotriester monomer units.[105] The ratio of monomer units was determined by the reaction time and the solvent (Scheme 24), the maximum MW was 2.3 kDa.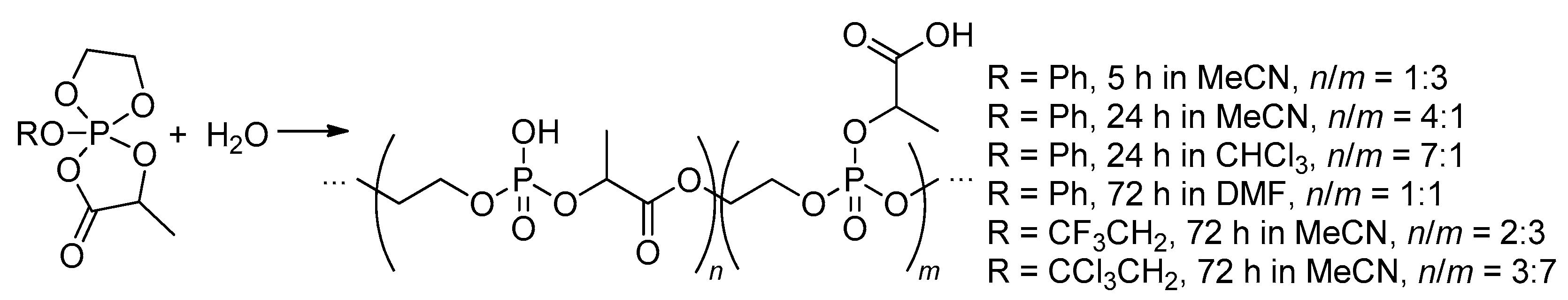
6.4. Thiol-ene Polyaddition
Recently Wurm and coll. proposed a new approach to PCPAs based on metal-free-radical thiol-ene polyaddition of dithiol comonomer and bis(alkenyl) phosphate to produce alternating copolymer with hydrophilic ethylene glycol segments in the polymer backbone (Scheme 25). To increase the hydrophilicity of the polymer, it was oxidized to the sulfone.[106]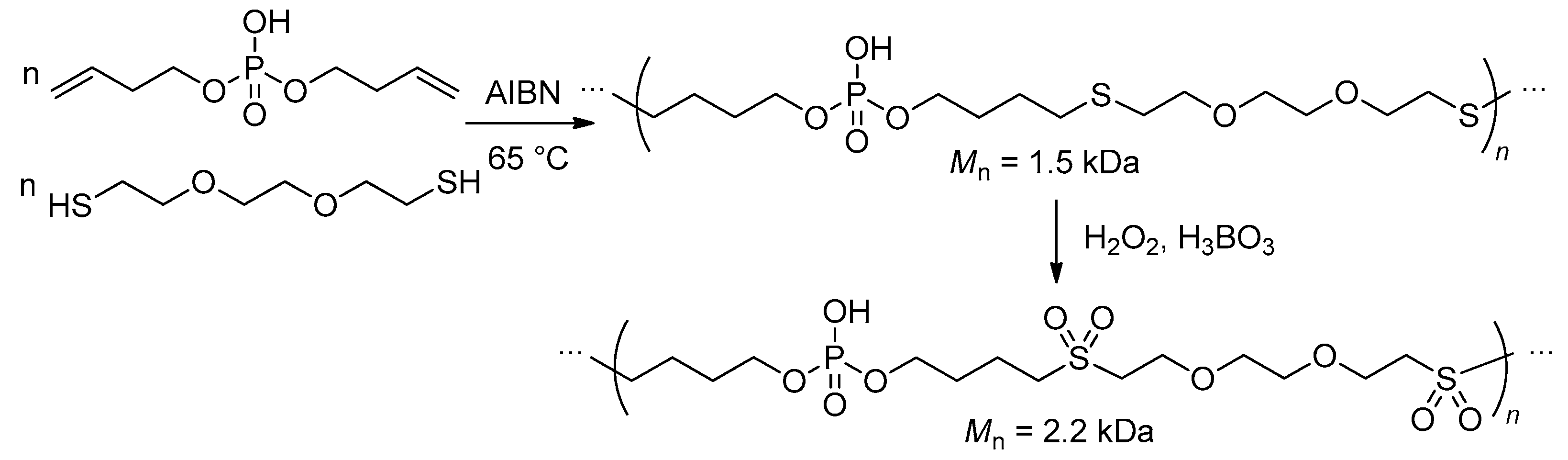
Scheme 25. Preparation of PCPAs via thiol-ene polyaddition and subsequent oxidation by hydrogen peroxide.[106]
6.5. Chain-End Vinyl Functionalization
Iwasaki described the use of methacrylamide-containing initiator in ROP of MeOEP, followed by the reaction with Me3N, to obtain functionalized Na-PEP (Scheme 26) suitable for free-radical graft polymerization[100]. Strictly speaking, the products of the latter reaction cannot easily be classified as ‘main’- or ‘side’-chain PCPAs, such attribution depends on the length of the grafted polymer.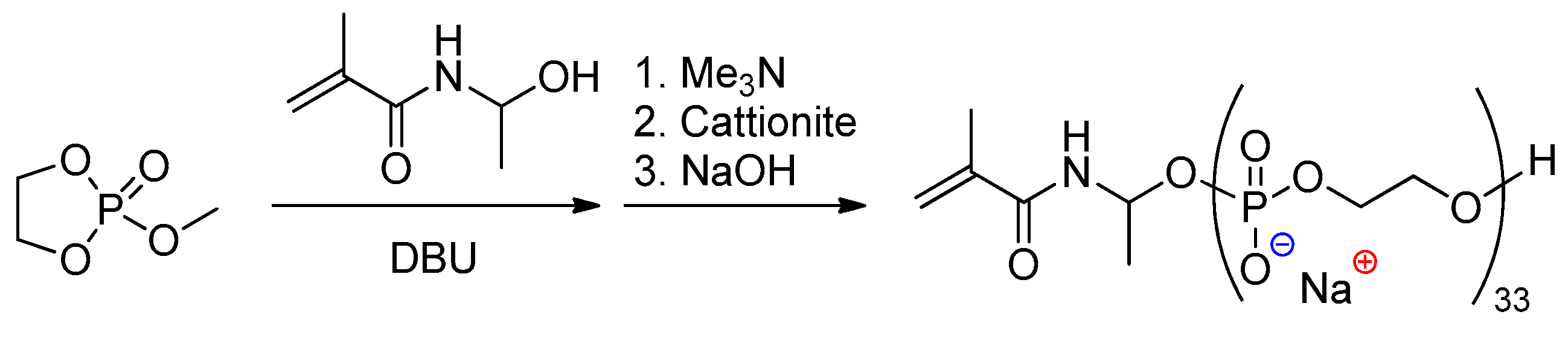
Scheme 26. Synthetic scheme of the polyphosphoester macromonomers.[100]
6.6. The Use of Bridged Cyclic Phosphates
Highly branched phosphate nanogels were obtained by polymerization of bridged cyclic phosphoester, 3,6-dioxaoctan-1,8-diyl bis(ethylene phosphate) and tris(2-aminoethyl)amine, in the presence of Triton X-100 in cyclohexane.[107] The product of this reaction contained three types of structural fragments (Scheme 27).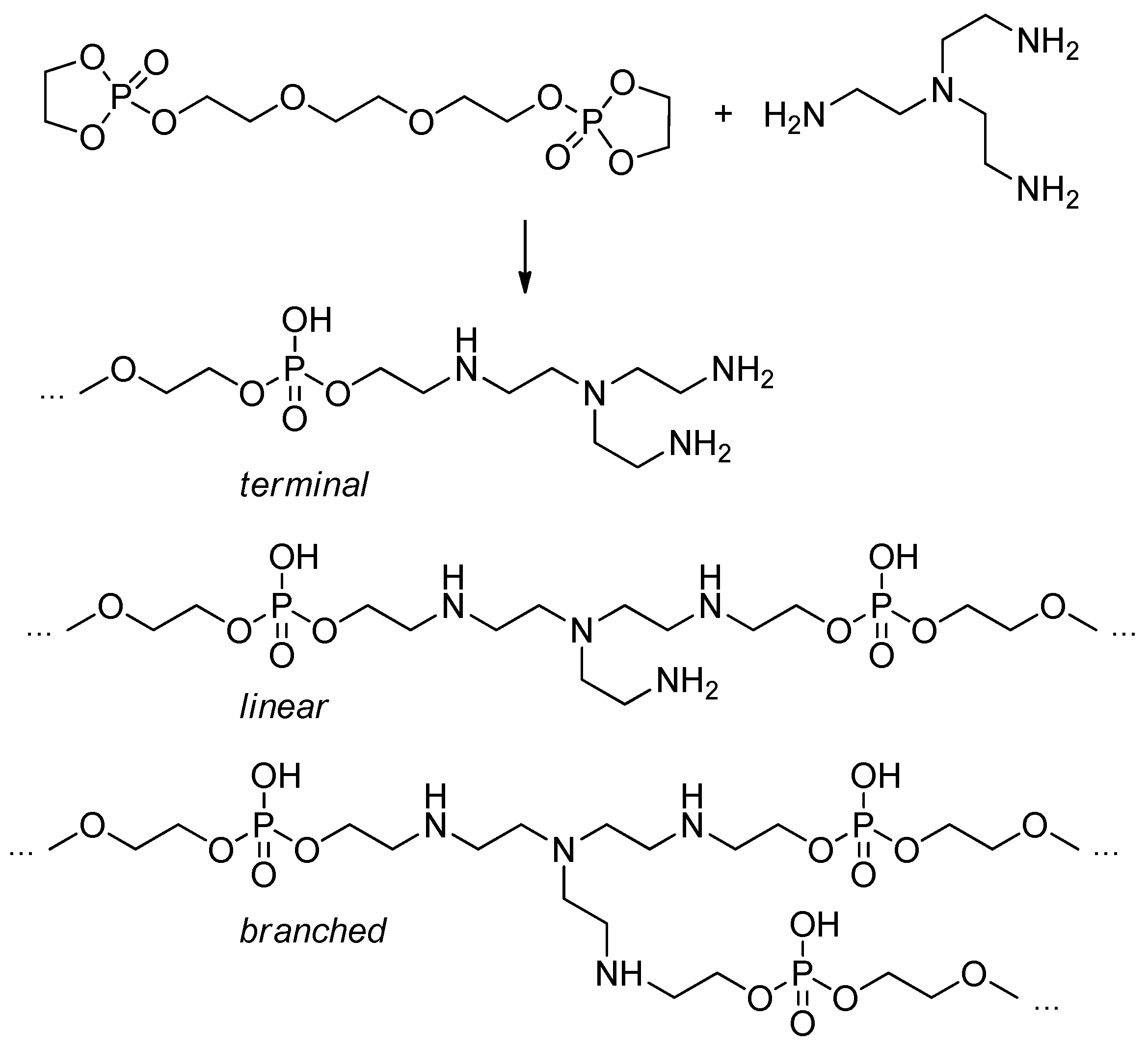
Scheme 27. Various structures of the reaction products of 3,6-dioxaoctan-1,8-diyl bis(ethylene phosphate) with tris(2-aminoethyl)amine.[107]
6.7. Post-Modification of Polyphosphodiesters
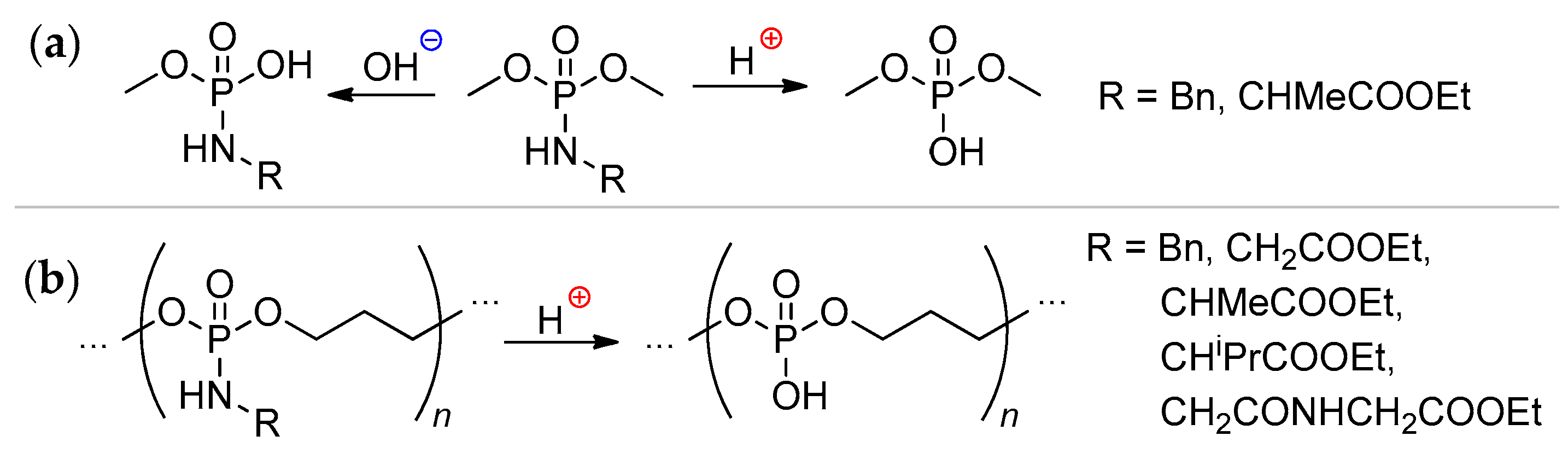
Polyphosphodiesters contain reactive acidic P–OH fragments and can of course be chemically modified. The reaction of PCPAs with oxirans (oxyethylation) stops when all of the acidic groups are consumed,[22] the synthesis of PEGylated polyphosphoesters requires the addition of an ‘external’ acid. Iwasaki synthesized polyphosphoester containing P–OCH2OAc and P–OH groups by the reaction of poly(EtOEP)-ran-PEPA with acetoxymethyl bromide.[69]7. Sequence-Defined Oligophosphodiesters
Nucleic acids are PCPAs that serve as the primary information-carrying molecules in cells. These natural PCPAs can be considered as sequence-defined poly(phosphodiesters) containing limited numbers of the ‘building blocks’. The maximum of the researchers’ interests in this area was highest during the 1980s, organochemical approaches to artificial DNA and close DNA analogs had been reviewed by Caruthers in 1991.[102] The synthesis of ‘artificial’ nucleic acids is based on ‘phosphoramidite’ chemistry (Scheme 28),[108][109] initially developed for solid-phase DNA synthesis.[102]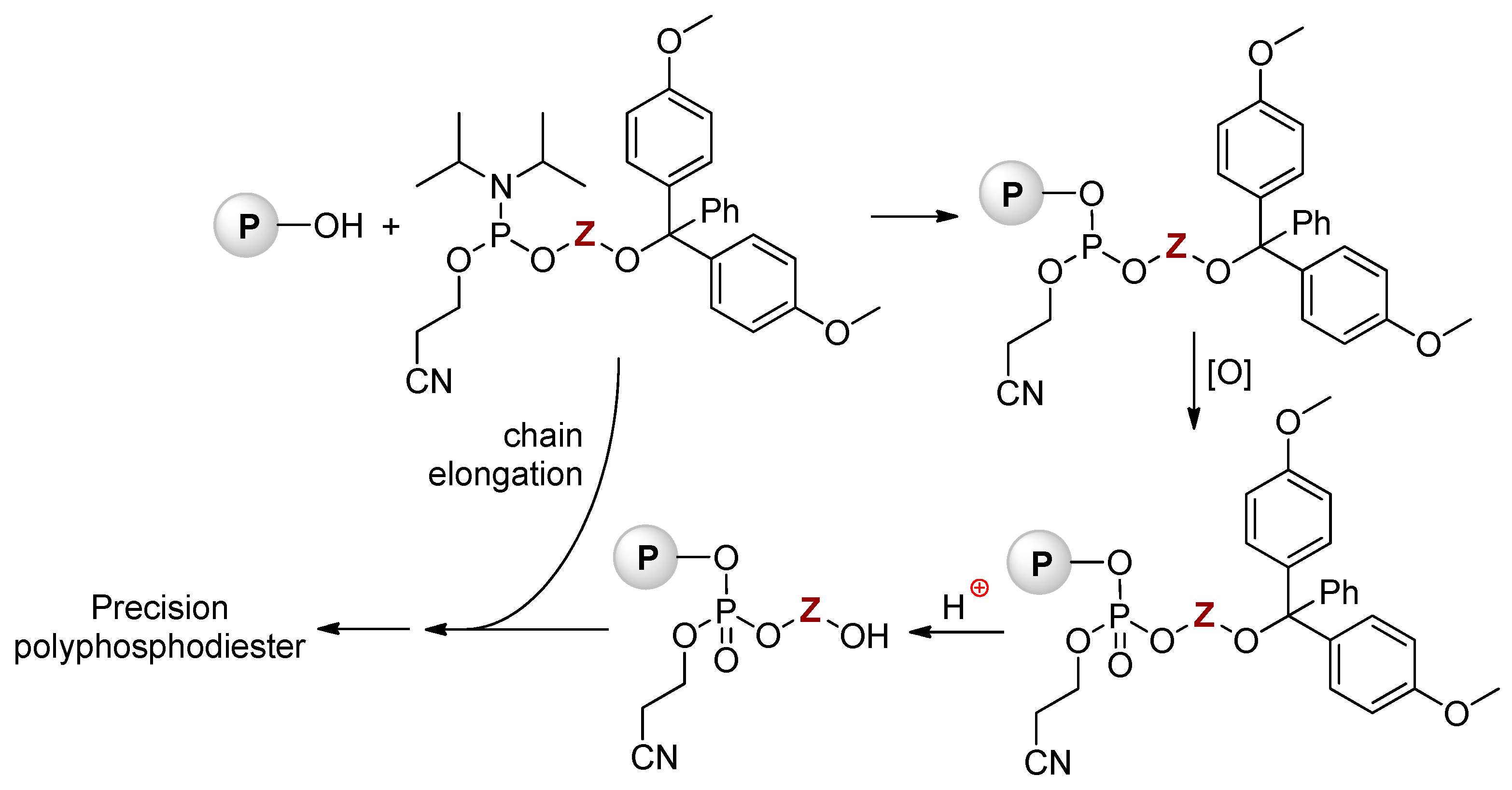
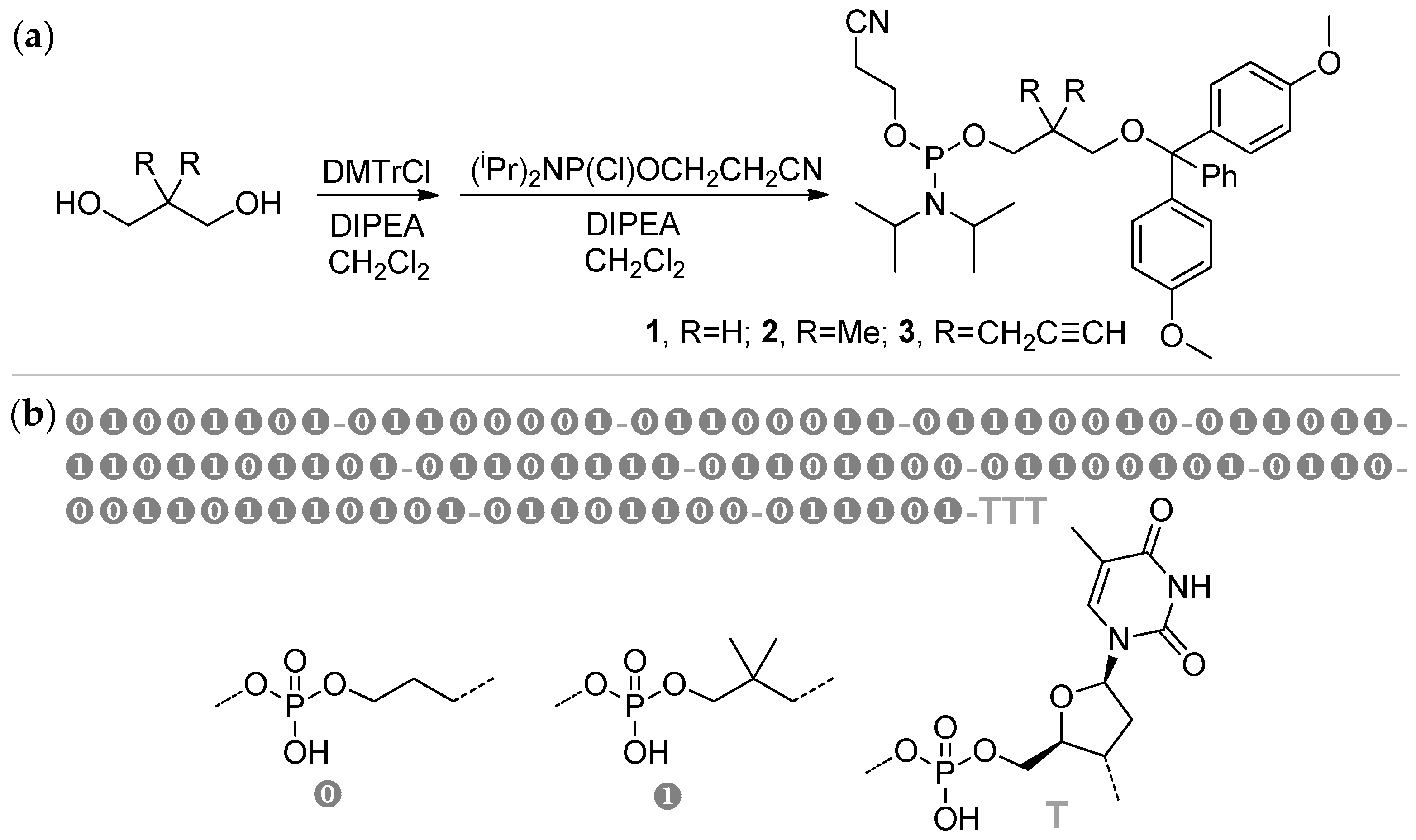
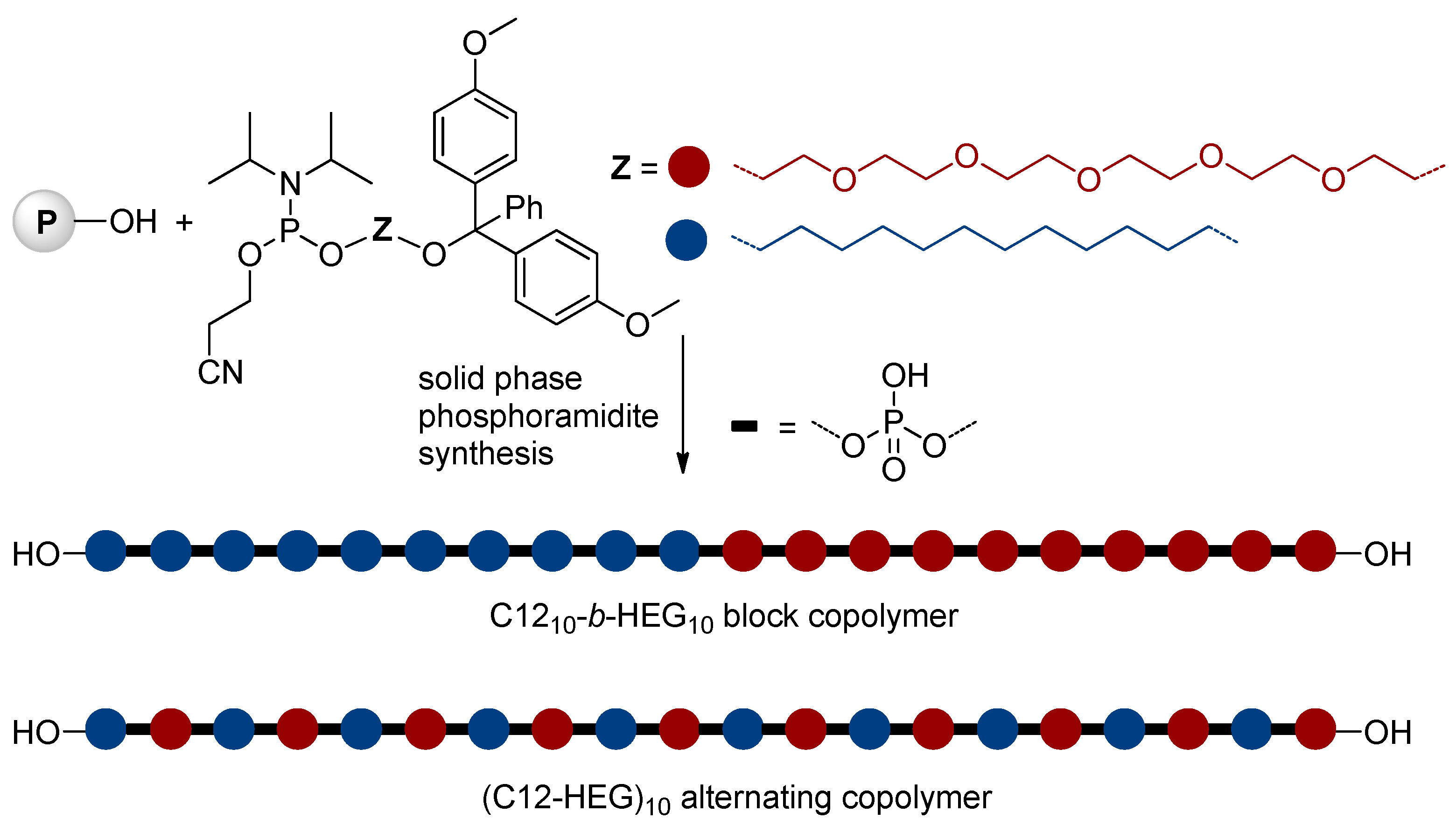
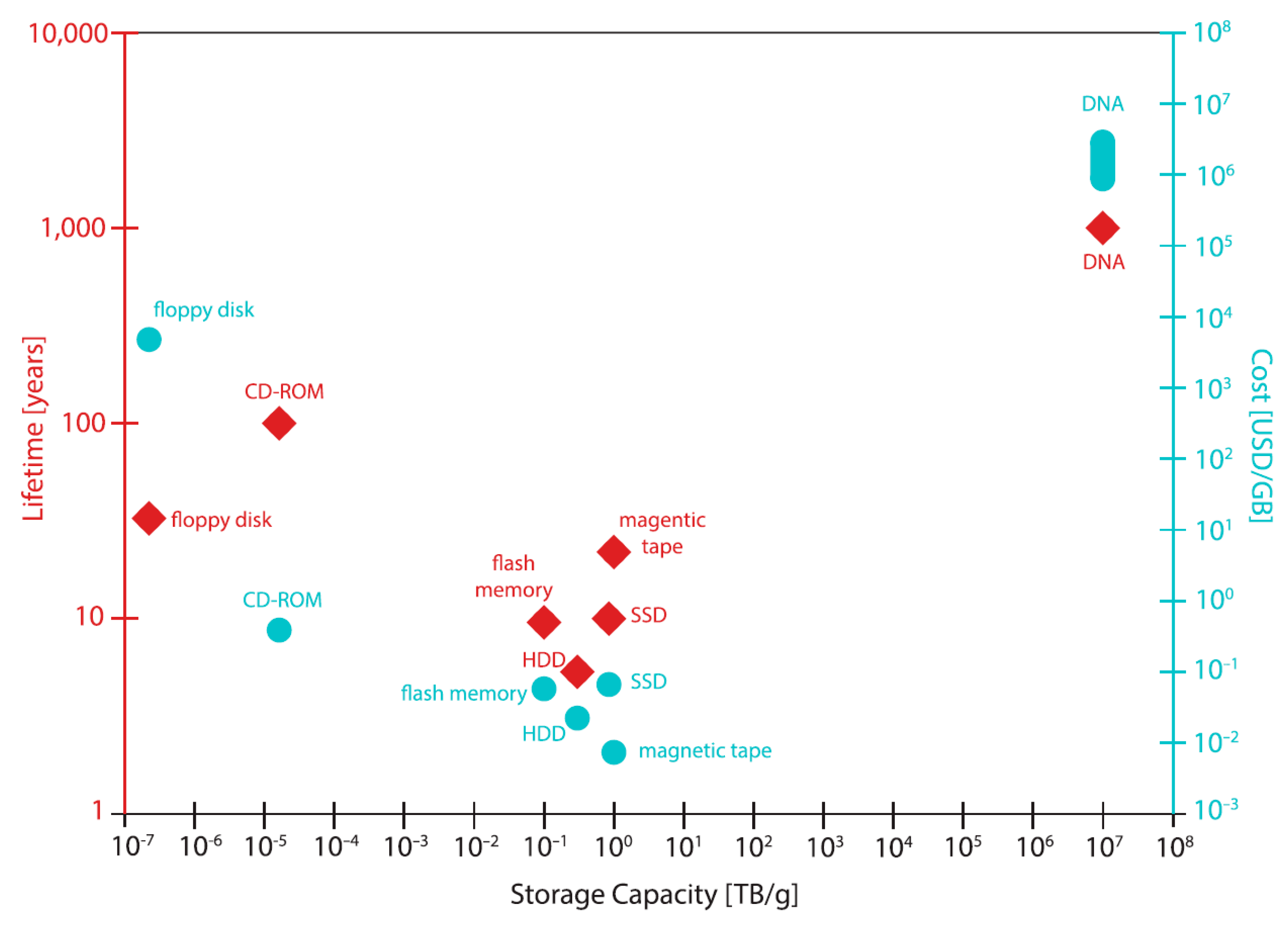
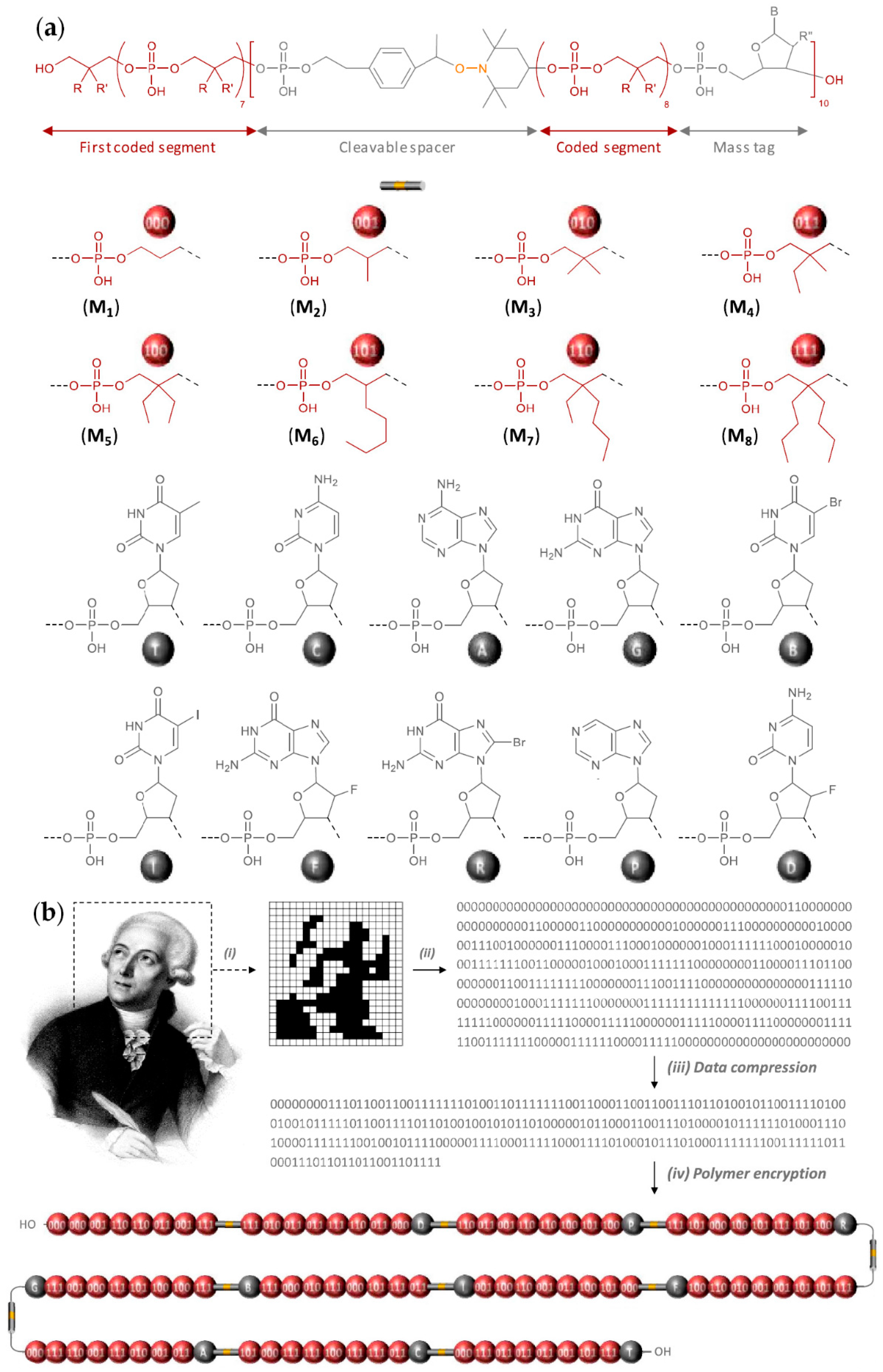
8. Conclusion
References
- Yilmaz, Z.E.; Jérôme, C.; Polyphosphoesters: New trends in synthesis and drug delivery applications. Macromol. Biosci. 2016, 16, 2210–2226, 10.1002/mabi.201600269.
- Appukutti, N.; Serpell, C.J.; High definition polyphosphoesters: Between nucleic acids and plastics. Polym. Chem. 2018, 9, 2210–2226, 10.1039/c8py00251g.
- Yilmaz, Z.E.; Jérôme, C.; Polyphosphoesters: New trends in synthesis and drug delivery applications. Macromol. Biosci. 2016, 16, 2210–2226, 10.1002/mabi.201600269.
- Bauer, K.N.; Tee, H.T.; Velencoso, M.M.; Wurm, F.R.; Main-chain poly(phosphoester)s: History, syntheses, degradation, bio- and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122, 10.1016/j.progpolymsci.2017.05.004.
- Iwasaki, Y.; Bone Mineral Affinity of Polyphosphodiesters. Molecules 2020, 55, 758, 10.3390/molecules25030758.
- Penczek, S.; Pretula, J.; Kaluzynski, K.; Poly(alkylene phosphates): From Synthetic Models of Biomacromolecules and Biomembranes toward Polymer−Inorganic Hybrids (Mimicking Biomineralization). Biomacromolecules 2005, 6, 547–551, 10.1021/bm049437f.
- Penczek, S.; Pretula, J.B.; Kaluzynski, K.; Lapienis, G.; Polymers with Esters of Phosphoric Acid Units: From Synthesis, Models of Biopolymers to Polymer—Inorganic Hybrids. Isr. J. Chem. 2012, 52, 306–319, 10.1002/ijch.201100162.
- Müller, W.E.G.; Schröder, H.C.; Wang, X.; Inorganic Polyphosphates As Storage for and Generator of Metabolic Energy in the Extracellular Matrix. Chem. Rev. 2019, 119, 12337–12374, 10.1021/acs.chemrev.9b00460.
- Mao, C.; The Emergence of Complexity: Lessons from DNA. PLoS Biol. 2004, 2, e431, 10.1371/journal.pbio.0020431.
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S.; Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2010, 11, 35–45, 10.1002/cbic.200900557.
- Steinbach, T.; Wurm, F.R.; Poly(phosphoester)s: A new platform for degradable polymers. Angew. Chem. Int. Ed. 2015, 54, 6098–6108, 10.1002/anie.201500147.
- Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J.; Phosphorus-Containing Polymers: A Great Opportunity for the Biomedical Field. Biomacromolecules 2011, 12, 1973–1982, 10.1021/bm2004803.
- Strasser, P.; Teasdale, I.; Main-Chain Phosphorus-Containing Polymers for Therapeutic Applications. Molecules 2020, 25, 1716, 10.3390/molecules25071716.
- Pelosi, C.; Tinè, M.R.; Wurm, F.R.; Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. 2020, 141, 110079, 10.1016/j.eurpolymj.2020.110079.
- Gao, X.; Li, L.; Cai, X.; Huang, Q.; Xiao, J.; Cheng, Y.; Targeting nanoparticles for diagnosis and therapy of bone tumors: Opportunities and challenges. Biomaterials 2021, 265, 120404, 10.1016/j.biomaterials.2020.120404.
- Hiranphinyophat, S.; Iwasaki, Y.; Controlled biointerfaces with biomimetic phosphorus-containing polymers. Sci. Technol. Adv. Biomater. 2021, 22, 301–316, 10.1080/14686996.2021.1908095.
- Wurm, F.R.; Binding matters: Binding patterns control the degradation of phosphorus-containing polymers. Green Mater. 2016, 4, 135–139, 10.1680/jgrma.16.00016.
- Lu, S.-Y.; Hamerton, I.; Recent developments in the chemistry of halogen-free flame retardant polymers. Prog. Polym. Sci. 2002, 27, 1661–1712, 10.1016/s0079-6700(02)00018-7.
- Jones, A.S.; Synthetic analogues of nucleic acids – a review. Int. J. Biol. Macromol. 1979, 1, 194–207, 10.1016/0141-8130(79)90013-8.
- Kashida, H.; Murayama, K.; Asanuma, H.; Acyclic artificial nucleic acids with phosphodiester bonds exhibit unique functions. Polym. J. 2016, 48, 781–786, 10.1038/pj.2016.39.
- Kaluzynski, K.; Libisowski, J.; Penczek, S.; A New Class of Synthetic Polyelectrolytes. Acidic Polyesters of Phosphoric Acid (Poly(hydroxyalkylene phosphates)). Macromolecules 1976, 9, 365–367, 10.1021/ma60050a039.
- Penczek, S.; Pretula, J.; High-molecular-weight poly(alkylene phosphates) and preparation of amphiphilic polymers thereof. Macromolecules 1993, 26, 2228–2233, 10.1021/ma00061a014.
- Pretula, J.; Penczek, S.; Poly(ethylene glycol) ionomers with phosphate diester linkages. Makromol. Chem. Rapid. Commun. 1988, 9, 731–737, 10.1002/marc.1988.030091103.
- Mitova, V.; Slavcheva, S.; Shestakova, P.; Momekova, D.; Stoyanov, N.; Momekov, G.; Troev, K.; Koseva, N.; Polyphosphoester conjugates of dinuclear platinum complex: Synthesis and evaluation of cytotoxic and the proapoptotic activity. Eur. J. Med. Chem. 2014, 72, 127–136, 10.1016/j.ejmech.2013.11.014.
- Troev, K.; Naruoka, A.; Terada, H.; Kikuchi, A.; Makino, K.; New Efficient Method of Oxidation of Poly(alkylene-H-phosphonate)s: A Promising Route to Novel co-Polyphosphoesters. Macromolecules 2012, 45, 5698–5703, 10.1021/ma3011608.
- Troev, K.; Tsatcheva, I.; Koseva, N.; Georgieva, R.; Gitsov, I.; Immobilization of aminothiols on poly(oxyethylene H-phosphonate)s and poly(oxyethylene phosphate)s—An approach to polymeric protective agents for radiotherapy of cancer. J. Polym. Sci. A Polym. Chem. 2007, 45, 1349–1363, 10.1002/pola.21906.
- Wódzki, R.; Świa̧tkowski, M.; Pretula, J.; Kałużyñski, K.; Poly[poly(oxypropylene) phosphate] macroionophores for transport and separation of cations in a hybrid: Cation-exchange polymer and liquid membrane system. J. Appl. Polym. Sci. 2004, 93, 1436–1445, 10.1002/app.20585.
- Ilia, G.; Simulescu, V.; Mak, C.A.; Crasmareanu, E.; The Use of Transesterification Method for Obtaining Phosphorus-Containing Polymers. Adv. Polym. Technol. 2014, 33, 21437, 10.1002/adv.21437.
- Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S.; Living ring-opening polymerizations of heterocyclic monomers. Prog. Polym. Sci. 2007, 32, 247–282, 10.1016/j.progpolymsci.2007.01.002.
- Becker, G.; Wurm, F.R.; Functional biodegradable polymers via ring-opening polymerization of monomers without protective groups. Chem. Soc. Rev. 2018, 47, 7739–7782, 10.1039/c8cs00531a.
- Steinmann, M.; Markwart, J.; Wurm, F.R.; Poly(alkylidene chlorophosphate)s via Acyclic Diene Metathesis Polymerization: A General Platform for the Postpolymerization Modification of Poly(phosphoester)s. Macromolecules 2014, 47, 8506–8513, 10.1021/ma501959h.
- Becker, G.; Ackermann, L.-M.; Schechtel, E.; Klapper, M.; Tremel, W.; Wurm, F.R.; Joining Two Natural Motifs: Catechol-Containing Poly(phosphoester)s. Biomacromolecules 2017, 18, 767–777, 10.1021/acs.biomac.6b01613.
- Tee, H.T.; Koynov, K.; Reichel, T.; Wurm, F.R.; Noncovalent Hydrogen Bonds Tune the Mechanical Properties of Phosphoester Polyethylene Mimics. ACS Omega 2019, 4, 9324–9332, 10.1021/acsomega.9b01040.
- Markwart, J.C.; Wurm, F.R.; The 2-acetylthioethyl ester group: A versatile protective group for P-OH-groups. Tetrahedron 2018, 74, 7426–7430, 10.1016/j.tet.2018.11.002.
- Markwart, J.C.; Suraeva, O.; Haider, T.; Lieberwirth, I.; Graf, R.; Wurm, F.R.; Defect engineering of polyethylene-like polyphosphoesters: Solid-state NMR characterization and surface chemistry of anisotropic polymer nanoplatelets. Polym. Chem. 2020, 11, 7235–7243, 10.1039/d0py01352h.
- Schulz, M.D.; Wagener, K.B.; Precision Polymers through ADMET Polymerization. Macromol. Chem. Phys. 2014, 215, 1936–1945, 10.1002/macp.201400268.
- Pretula, J.; Kaluzynski, K.; Wisniewski, B.; Szymanski, R.; Loontjens, T.; Penczek, S.; Formation of poly(ethylene phosphates) in polycondensation of H3PO4 with ethylene glycol. Kinetic and mechanistic study. J. Polym. Sci. A Polym. Chem. 2008, 46, 830–843, 10.1002/pola.22427.
- Pretula, J.; Kaluzynski, K.; Wisniewski, B.; Szymanski, R.; Loontjens, T.; Penczek, S.; H3PO4 in a direct synthesis of oligo–poly(ethylene phosphate)from ethylene glycol. J. Polym. Sci. Part A. Polym. Chem. 2006, 44, 2358–2362, 10.1002/pola.21332.
- Penczek, S.; Kaluzynski, K.; Pretula, J.; Phosphorylation of Polyols with H3PO4: Towards Simple Synthesis of Poly(alkylene phosphate)s. Phosph. Sulfur Silicon Rel. Elem. 2009, 184, 1935–1945, 10.1080/10426500903110017.
- Pretula, J.; Kaluzynski, K.; Szymanski, R.; Penczek, S.; Polycondensation of H3PO4 with glycerol: From branched structures to hydrolytically reversible gels. J. Polym. Sci. A Polym. Chem. 2014, 52, 3533–3542, 10.1002/pola.27421.
- Pretula, J.; Kaluzynski, K.; Penczek, S.; Polycondensation of diglycerol with H3PO4. Reversibly degradable gels giving multireactive, highly branched macromolecules. J. Polym. Sci. A Polym. Chem. 2016, 54, 3303–3317, 10.1002/9781118967904.
- Munoz, A.; Vives, J.P.; Petit, J.; On the polycindensation of phosphoric acid and cyclic ethylene glycol carbonate. C. R. Acad. Sci. 1963, 257, 1863–1866, 10.3390/ijms232314857.
- Imoto, M.; Ouchi, T.; Sakae, M.; Yamamoto, H.; Vinyl polymerization, 367. Polymerization of methyl methacrylate initiated with sodium polyethylenephosphate in the presence of an aqueous solution of copper(II) chloride. Macromol. Chem. Phys. 1980, 181, 341–349, 10.1002/macp.1980.021810205.
- Biela, T.; Szymanski, R.; Kubisa, P.; Oligomerization of oxiranes in the presence of phosphorus acids, 2. Kinetics of addition of ethylene oxide to phosphoric and phosphorous acid. Makromol. Chem. 1992, 193, 285–301, 10.1002/macp.1992.021930126.
- Biela, T.; Nyk, A.; Kubisa, P.; Polyphosphate chains by addition of oxiranes to phosphoric acid. Makromol. Chem. Macromol. Symp. 1992, 60, 155–163, 10.1002/masy.19920600114.
- Imran, M.; Kim, B.-K.; Han, M.; Cho, B.G.; Kim, D.H.; Sub- and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis(2-hydroxyethyl) terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693, 10.1016/j.polymdegradstab.2010.05.026.
- Ghasemi, M.H.; Neekzad, N.; Ajdari, F.B.; Kowsari, E.; Ramakrishna, S.; Mechanistic aspects of poly(ethylene terephthalate) recycling–toward enabling high quality sustainability decisions in waste management. Environ. Sci. Poll. Res. 2021, 28, 43074–43101, 10.1007/s11356-021-14925-z.
- Wan, A.C.A.; Mao, H.-Q.; Wang, S.; Phua, S.H.; Lee, G.P.; Pan, J.; Lu, S.; Wang, J.; Leong, K.W.; Poly(phosphoester) ionomers as tissue-engineering scaffolds. J. Biomed. Mater. Res. B: Appl. Biomat. 2004, 70, 91–102, 10.1002/jbm.b.30022.
- Klosinski, P.; Penczek, S.; Synthesis of models of teichoic acids by ring-opening polymerization. Macromolecules 1983, 16, 316–320, 10.1021/ma00236a029.
- Pretula, J.; Penczek, S.; High-molecular-weight poly(alkylene phosphonate)s by condensation of dialkylphosphonates with diols. Makromol. Chem. 1990, 191, 671–680, 10.1002/macp.1990.021910322.
- Pretula, J.; Kaluzynski, K.; Szymanski, R.; Penczek, S.; Transesterification of oligomeric dialkyl phosphonates, leading to the high-molecular-weight poly-H-phosphonates. J. Polym. Sci. A Polym. Chem. 1999, 37, 1365–1381, 10.1002/(SICI)1099-0518(19990501)37:9<1365::AID-POLA17>3.0.CO;2-%23.
- Penczek, S.; Pretula, J.; Kaluzynski, K.; Synthesis of a triblock copolymer: Poly(ethylene glycol)-poly(alkylene phosphate)-poly(ethylene glycol) as a modifier of CaCO3 crystallization. J. Polym. Sci. A Polym. Chem. 2005, 43, 650–657, 10.1002/pola.20525.
- Pretula, J.; Kaluzynski, K.; Szymanski, R.; Penczek, S.; Preparation of Poly(alkylene H-phosphonate)s and Their Derivatives by Polycondensation of Diphenyl H-Phosphonate with Diols and Subsequent Transformations. Macromolecules 1997, 30, 8172–8176, 10.1021/ma970390i.
- Kaluzynski, K.; Penczek, S.; Amino acids attached to poly(alkylene phosphate)s, 1. Synthesis. Macromol. Chem. Phys. 1994, 195, 3855–3862, 10.1002/macp.1994.021951211.
- Penczek, S.; Kaluzynski, K.; Baran, J.. Amino acid couples to poly(alkylene phosphates); Kahovec J., Eds.; VSP International Science Publishers: Utrecht, The Netherlands, 1993; pp. 231–240.
- Baran, J.; Kaluzynski, K.; Szymanski, R.; Penczek, S.; Hydrolysis of Poly(alkylene amidophosphate)s Containing Amino Acid or Peptide Residues in the Side Groups. Kinetics and Selectivity of Hydrolysis. Biomacromolecules 2004, 5, 1841–1848, 10.1021/bm0498171.
- Busch, H.; Schiebel, E.; Sickinger, A.; Mecking, S.; Ultralong-Chain-Spaced Crystalline Poly(H-phosphonate)s and Poly(phenylphosphonate)s. Macromolecules 2017, 50, 7901–7910, 10.1021/acs.macromol.7b01368.
- Kaluzynski, K.; Pretula, J.; Lewinski, P.; Kaźmierski, S.; Penczek, S.; Catalysis in polymerization of cyclic esters. Catalyst and initiator in one molecule. Polymerization of ε-caprolactone. J. Catal. 2020, 392, 97–107, 10.1016/j.jcat.2020.09.026.
- Kaluzynski, K.; Pretula, J.; Lewinski, P.; Kaźmierski, S.; Penczek, S.; Synthesis and Properties of Functionalized Poly(ε-caprolactone); Chain Polymerization Followed by Polycondensation in One Pot with Initiator and Catalyst in One Molecule. Synthesis and Molecular Structures. Macromolecules 2022, 55, 2210–2221, 10.1021/acs.macromol.1c02325.
- Lucas, H.J.; Mitchell Jr., F.W.; Scully, C.N.; Cyclic Phosphites of Some Aliphatic Glycols. J. Am. Chem. Soc. J. Am. Chem. Soc. , 72, 5491–5497, 10.1021/ja01168a032.
- Nifant'ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V.; Controlled ring-opening polymerisation of cyclic phosphates, phosphonates and phosphoramidates catalysed by hereroleptic BHT-alkoxy magnesium complexes. Polym. Chem. 2017, 8, 6806–6816, 10.1039/c7py01472d.
- Oussadi, K.; Montembault, V.; Belbachir, M.; Fontaine, L.; Ring-opening bulk polymerization of five- and six-membered cyclic phosphonates using maghnite, a nontoxic proton exchanged montmorillonite clay. J. Appl. Polym. Sci. 2011, 122, 891–897, 10.1002/app.34193.
- Maffei, M.; Buono, G.; A two step synthesis of 2-oxo-2-vinyl 1,3,2-dioxaphospholanes and -dioxaphosphorinanes. Tetrahedron 2003, 59, 8821–8825, 10.1016/j.tet.2003.08.067.
- Biela, T.; Penczek, S.; Slomkowski, S.; Vogl, O.; Racemic and optically active poly(4-methyl-2-oxo-2-hydro-1,3,2-dioxaphospholane). Synthesis and oxidation to the polyacids. Makromol. Chem. Rapid Commun. 1982, 3, 667–671, 10.1002/marc.1982.030031003.
- Lapienis, G.; Penczek, S.; Cationic Polymerization of 2-Alkoxy-2-oxo-1,3,2-dioxaphosphorinanes (1,3-Propylene Alkyl Phosphates). Macromolecules 1977, 10, 1301–1306, 10.1021/ma60060a027.
- Libiszowski, J.; Kałużynski, K.; Penczek, S.; Polymerization of cyclic esters of phosphoric acid. VI. Poly(alkyl ethylene phosphates). Polymerization of 2-alkoxy-2-oxo-1,3,2-dioxaphospholans and structure of polymers. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 1275–1283, 10.1002/pol.1978.170160610.
- Becker, G.; Wurm, F.R.; Breathing air as oxidant: Optimization of 2-chloro-2-oxo-1,3,2-dioxaphospholane synthesis as a precursor for phosphoryl choline derivatives and cyclic phosphate monomers. Tetrahedron 2017, 73, 3536–3540, 10.1016/j.tet.2017.05.037.
- Morodo, R.; Riva, R.; van den Akker, N.M.S.; Molin, D.G.M.; Jérôme, C.; Monbaliu, J.-C.M.; Accelerating the end-to-end production of cyclic phosphate monomers with modular flow chemistry. Chem. Sci. 2022, 13, 10699–10706, 10.1039/d2sc02891c.
- Iwasaki, Y.; Kawakita, T.; Yusa, S.; Thermoresponsive Polyphosphoesters Bearing Enzyme-cleavable Side Chains. Chem. Lett. 2009, 38, 1054–1055, 10.1246/cl.2009.1054.
- Yasuda, H.; Sumitani, M.; Nakamura, A.; Novel Synthesis of Acidic Polyesters of Phosphoric Acid by Thermal Elimination of Isobutylene from Poly(alkylene tert-butyl phosphates). Macromolecules 1981, 14, 458–460, 10.1021/ma50003a046.
- Lapienis, G.; Penczek, S.; Pretula, J.; Poly (dialkylphosphates) based on deoxyribose. Macromolecules 1983, 16, 153–158, 10.1021/ma00236a001.
- Olsén, P.; Odelius, K.; Albertsson, A.-C.; Thermodynamic Presynthetic Considerations for Ring-Opening Polymerization. Biomacromolecules 2016, 17, 699–709, 10.1021/acs.biomac.5b01698.
- Sosnowski, S.; Libiszowski, J.; Słomkowski, S.; Penczek, S.; Thermodynamics of the polymerization of ethylene methyl phosphate. Makromol. Chem. Rapid Commun. 1984, 5, 239–244, 10.1002/marc.1984.030050501.
- Penczek, S.; Mechanism of ionic polymerization of cyclic esters of phosphoric acid (a new route to models of biopolymers). J. Polym. Sci. Polym. Symp. 1980, 67, 149–168, 10.1002/polc.5070670111.
- Penczek, S.; Biela, T.; Klosinski, P.; Lapienis, G.; Polymerization of phosphorus containing cyclic monomers: Synthesis of polymers related to biopolymers. Makromol. Chem., Macromol. Symp. 1986, 6, 123–153, 10.1002/masy.19860060114.
- Pretula, J.; Kałużyṅski, K.; Penczek, S.; Living reversible anonic polymerization of N ,N--diethylamine-1,3,2-dioxaphosphorinan. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 1251–1258, 10.1002/pol.1984.170220605.
- Bauer, K.N.; Liu, L.; Wagner, M.; Andrienko, D.; Wurm, F.R.; Mechanistic study on the hydrolytic degradation of polyphosphates. Eur. Polym. J. 2018, 108, 286–294, 10.1016/j.eurpolymj.2018.08.05.
- Nifantev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Kosarev, M.A.; Gavrilov, D.E.; Komarov, P.D.; Ilyin, S.O.; Karchevsky, S.G.; Ivchenko, P.V.; Mechanistic study of transesterification in TBD-catalyzed ring-opening polymerization of methyl ethylene phosphate. Eur. Polym. J. 2019, 118, 393–403, 10.1016/j.eurpolymj.2019.06.015.
- Hirano, Y.; Iwasaki, Y.; Bone-specific poly(ethylene sodium phosphate)-bearing biodegradable nanoparticles. Coll. Surf. B: Biointerfaces 2017, 153, 104–110, 10.1016/j.colsurfb.2017.02.015.
- Noree, S.; Iwasaki, Y.; Thermally Assisted Generation of Protein–Poly(ethylene sodium phosphate) Conjugates with High Mineral Affinity. ACS Omega 2019, 4, 3398–3404, 10.1021/acsomega.8b03585.
- Otaka, A.; Iwasaki, Y.; Endocytosis of poly(ethylene sodium phosphate) by macrophages and the effect of polymer length on cellular uptake. J. Ind. Eng. Chem. 2019, 75, 115–122, 10.1016/j.jiec.2019.03.010.
- Iwasaki, Y.; Yokota, A.; Otaka, A.; Inoue, N.; Yamaguchi, A.; Yoshitomi, T.; Yoshimotode, K.; Neo, M.; Bone-targeting poly(ethylene sodium phosphate). Biomater. Sci. 2018, 6, 91–95, 10.1039/c7bm00930e.
- Clément, B.; Molin, D.G.; Jérôme, C.; Lecomte, P.; Synthesis of polyphosphodiesters by ring-opening polymerization of cyclic phosphates bearing allyl phosphoester protecting groups. J. Polym. Sci. A Polym. Chem. 2015, 53, 2642–2648, 10.1002/pola.27732.
- Ergul Yilmaz, Z.; Debuigne, A.; Calvignac, B.; Boury, F.; Jérôme, C.; Double hydrophilic polyphosphoester containing copolymers as efficient templating agents for calcium carbonate microparticles. J. Mater. Chem. B 2015, 3, 7227–7236, 10.1039/c5tb00887e.
- Nifantev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Tavtorkin, A.N.; Minyaev, M.E.; Kosarev, M.A.; Ivchenko, P.V.; Synthesis in aqueous media of poly(ethylene phosphoric acids) by mild thermolysis of homopolymers and block copolymers based on tert-butyl ethylene phosphate. Eur. Polym. J. 2018, 106, 249–256, 10.1016/j.eurpolymj.2018.07.034.
- Nifant’ev, I.; Siniavin, A.; Karamov, E.; Kosarev, M.; Kovalchuk, S.; Turgiev, A.; Nametkin, S.; Bagrov, V.; Tavtorkin, A.; Ivchenko, P.; et al. A New Approach to Developing Long-Acting Injectable Formulations of Anti-HIV Drugs: Poly(Ethylene Phosphoric Acid) Block Copolymers Increase the Efficiency of Tenofovir against HIV-1 in MT-4 Cells. Int. J. Mol. Sci. 2021, 22, 340, 10.3390/ijms22010340.
- Iwasaki, Y.; Katayama, K.; Yoshida, M.; Yamamoto, M.; Tabata, Y.; Comparative physicochemical properties and cytotoxicity of polyphosphoester ionomers with bisphosphonates. J. Biomater. Sci. Polym. Ed. 2012, 24, 882–895, 10.1080/09205063.2012.710823.
- Ikeuchi, R.; Iwasaki, Y.; High mineral affinity of polyphosphoester ionomer–phospholipid vesicles. J. Biomed. Mater. Res. 2013, 101, 318–325, 10.1002/jbm.a.34321.
- Zhang, S.; Wang, H.; Shen, Y.; Zhang, F.; Seetho, K.; Zou, J.; Taylor, J.-S.A.; Dove, A.P.; Wooley, K.L.; A simple and efficient synthesis of an acid-labile polyphosphoramidate by organobase-catalyzed ring-opening polymerization and transformation to polyphosphoester ionomers by acid treatment. Macromolecules 2013, 46, 5141–5149, 10.1021/ma400675m.
- Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Karchevsky, S.; Ivchenko, P.; Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers Polymers , 10, 1105, 10.3390/polym10101105.
- Nifant’ev, I.; Gavrilov, D.; Tavtorkin, A.; Chinova, M.; Besprozvannykh, V.; Komarov, P.; Zaitsev, V.; Podoprigora, I.; Ivchenko, P.; Antibacterial Poly(ε-CL)/Hydroxyapatite Electrospun Fibers Reinforced by Poly(ε-CL)-b-poly(ethylene phosphoric acid). Int. J. Mol. Sci. 2021, 22, 7690, 10.3390/ijms22147690.
- Lapienis, G.. Ring-Opening Polymerization of Cyclic Phosphorus Monomers. In: Polymer Science: A Comprehensive Reference; Elsevier B.V.: Amsterdam, Netherlands, 2012; pp. 477–505.
- Kluger, R.; Taylor, S.D.; Endocyclic cleavage in the alkaline hydrolysis of the cyclic phosphonate methyl propylphostonate: Dianionic intermediates and barriers to pseudorotation. J. Am. Chem. Soc. 1991, 113, 5714–5719, 10.1021/ja00015a027.
- Wang, J.; Sun, D.D.N.; Shin-ya, Y.; Leong, K.W.; Stimuli-Responsive Hydrogel Based on Poly(propylene phosphate). Macromolecules 2004, 37, 670–672, 10.1021/ma035453d.
- Gehrmann, T.; Vogt, W.; Polymer ester von sären des phosphors, 7. Polymerisation des 1-oxo-2,6,7-trioxa-1-phosphabicyclo [2.2.1]heptans. Makromol. Chem. 1981, 182, 3069–3076, 10.1002/macp.1981.021821114.
- Baran, J.; Penczek, S.; Hydrolysis of Polyesters of Phosphoric Acid. 1. Kinetics and the pH Profile. Macromolecules 1995, 28, 5167–5176, 10.1021/ma00119a002.
- Yasuda, H.; Sumitani, M.; Lee, K.; Araki, T.; Nakamura, A.; High molecular weight poly(2-methoxy-1,3,2-dioxaphospholane 2-oxide) by ring-opening catalysis of tertiary amines. Initiation and stepwise propagation mechanisms as studied by the stoichiometric reaction with triethylamine. Macromolecul 1982, 15, 1231–1237, 10.1021/ma00233a003.
- Kootala, S.; Tokunaga, M.; Hilborn, J.; Iwasaki, Y.; Anti-Resorptive Functions of Poly(ethylene sodium phosphate) on Human Osteoclasts. Macromol. Biosci. 2015, 15, 1634–1640, 10.1002/mabi.201500166.
- Iwasaki, Y.; Takahata, Y.; Fujii, S.; Self-setting particle-stabilized emulsion for hard-tissue engineering. Colloids Surfaces B: Biointerfaces 2015, 126, 394–400, 10.1016/j.colsurfb.2014.12.003.
- Kunomura, S.; Iwasaki, Y.; Immobilization of polyphosphoesters on poly(ether ether ketone) (PEEK) for facilitating mineral coating. J. Biomater. Sci. Polym. Ed. 2019, 30, 861–876, 10.1080/09205063.2019.1595305.
- Li, R.; Elsabahy, M.; Song, Y.; Wang, H.; Su, L.; Letteri, R.A.; Khan, S.; Heo, G.S.; Sun, G.; Liu, Y.; et al.et al. Functional, Degradable Zwitterionic Polyphosphoesters as Biocompatible Coating Materials for Metal Nanostructures. Langmuir 2019, 35, 1503–1512, 10.1021/acs.langmuir.8b02033.
- Caruthers, M.H.; Chemical synthesis of DNA and DNA analogs. Acc. Chem. Res. 1991, 24, 278–284, 10.1021/ar00009a005.
- Dera, R.; Diliën, H.; Billen, B.; Gagliardi, M.; Rahimi, N.; Van Den Akker, N.M.S.; Molin, D.G.M.; Grandfils, C.; Adriaensens, P.; Guedens, W.; et al.et al. Phosphodiester Hydrogels for Cell Scaffolding and Drug Release Applications. Macromol. Biosci. 2019, 19, 1900090, 10.1002/mabi.201900090.
- Dera, R.; Diliën, H.; Adriaensens, P.; Guedens, W.; Cleij, T.J.; An Efficient Thermal Elimination Pathway toward Phosphodiester Hydrogels via a Precursor Approach. Macromol. Chem. Phys. 2020, 221, 1900466, 10.1002/macp.201900466.
- Kobayashi, S.; Narukawa, Y.; Saegusa, T.; Hydrolysis polymerization of spiro(acylpentaoxy)phosphoranes to polyphosphates. Macromolecules 1984, 17, 134–138, 10.1021/ma00132a004.
- Steinmann, M.; Wurm, F.R.; Water-soluble and degradable polyphosphorodiamidates via thiol-ene polyaddition. Polym. Degrad. Stab. 2020, 179, 109224, 10.1016/j.polymdegradstab.2020.109224.
- Yuan, Y.-Y.; Du, J.-Z.; Song, W.-J.; Wang, F.; Yang, X.-Z.; Xiong, M.-H.; Wang, J.; Biocompatible and functionalizable polyphosphate nanogel with a branched structure. J. Mater. Chem. 2012, 22, 9322–9329, 10.1039/c2jm30663h.
- Al Ouahabi, A.; Kotera, M.; Charles, L.; Lutz, J.-F.; Synthesis of Monodisperse Sequence-Coded Polymers with Chain Lengths above DP100. ACS Macro Lett. 2015, 4, 1077–1080, 10.1021/acsmacrolett.5b00606.
- Vybornyi, M.; Vyborna, Y.; Häner, R.; DNA-inspired oligomers: From oligophosphates to functional materials. Chem. Soc. Rev. 2019, 48, 4347–4360, 10.1039/c8cs00662h.
- Charles, L.; Lutz, J.-F.; Design of Abiological Digital Poly(phosphodiester)s. Acc. Chem. Res. 2021, 54, 1791–1800, 10.1021/acs.accounts.1c00038.
- Meiser, L.C.; Nguyen, B.H.; Chen, Y.-J.; Nivala, J.; Strauss, K.; Ceze, L.; Grass, R.N.; Synthetic DNA applications in information technology. Nat. Commun. 2022, 13, 352, 10.1038/s41467-021-27846-9.
- Al Ouahabi, A.; Charles, L.; Lutz, J.-F.; Synthesis of Non-Natural Sequence-Encoded Polymers Using Phosphoramidite Chemistry. J. Am. Chem. Soc. 2015, 137, 5629–5635, 10.1021/jacs.5b02639.
- Roszak, I.; Oswald, L.; Al Ouahabi, A.; Bertin, A.; Laurent, E.; Felix, O.; Carvin-Sergent, I.; Charles, L.; Lutz, J.-F.; Synthesis and sequencing of informational poly(amino phosphodiester)s. Polym. Chem. 2021, 12, 5279–5282, 10.1039/d1py01052b.
- Loth, C.; Charles, L.; Lutz, J.-F.; Nerantzaki, M.; Precisely Defined Aptamer-b-Poly(phosphodiester) Conjugates Prepared by Phosphoramidite Polymer Chemistry. ACS Macro Lett. 2021, 10, 481–485, 10.1021/acsmacrolett.1c00164.
- Laurent, E.; Amalian, J.-A.; Parmentier, M.; Oswald, L.; Al Ouahabi, A.; Dufour, F.; Launay, K.; Clément, J.-L.; Gigmes, D.; Delsuc, M.-A.; et al.et al. High-Capacity Digital Polymers: Storing Images in Single Molecules. Macromolecules 2020, 53, 4022–4029, 10.1021/acs.macromol.0c00666.
- de Rochambeau, D.; Sun, Y.; Barlog, M.; Bazzi, H.S.; Sleiman, H.F.; Modular Strategy To Expand the Chemical Diversity of DNA and Sequence-Controlled Polymers. J. Org. Chem. 2018, 83, 9774–9786, 10.1021/acs.joc.8b01184.
- Launay, K.; Amalian, J.-A.; Laurent, E.; Oswald, L.; Al Ouahabi, A.; Burel, A.; Dufour, F.; Carapito, C.; Clément, J.-L.; Lutz, J.-F.; et al.et al. Precise Alkoxyamine Design to Enable Automated Tandem Mass Spectrometry Sequencing of Digital Poly(phosphodiester)s. Angew. Chem. Int. Ed. 2021, 60, 917–926, 10.1002/anie.202010171.
- König, N.F.; Al Ouahabi, A.; Oswald, L.; Szweda, R.; Charles, L.; Lutz, J.-F.; Photo-editable macromolecular information. Nat. Commun. 2019, 10, 3774, 10.1038/s41467-019-11566-2.
- Appukutti, N.; Jones, J.R.; Serpell, C.J.; Sequence isomerism in uniform polyphosphoesters programmes self-assembly and folding. Chem. Commun. 2020, 56, 5307–5310, 10.1039/d0cc01319f.
- Amalian, J.-A.; Mondal, T.; Konishcheva, E.; Cavallo, G.; Petit, B.E.; Lutz, J.-F.; Charles, L.; Desorption Electrospray Ionization (DESI) of Digital Polymers: Direct Tandem Mass Spectrometry Decoding and Imaging from Materials Surfaces. Adv. Mater. Technol. 2021, 6, 2001088, 10.1002/admt.202001088.
- Amalian, J.-A.; Al Ouahabi, A.; Cavallo, G.; König, N.F.; Poyer, S.; Lutz, J.-F.; Charles, L.; Controlling the structure of sequence-defined poly(phosphodiester)s for optimal MS/MS reading of digital information. J. Mass Spectr. 2017, 52, 788–798, 10.1002/jms.3947.
- Laurent, E.; Amalian, J.-A.; Schutz, T.; Launay, K.; Clément, J.-L.; Gigmes, D.; Burel, A.; Carapito, C.; Charles, L.; Delsuc, M.-A.; et al.et al. Storing the portrait of Antoine de Lavoisier in a single macromolecule. Compt. Rend. Chim. 2021, 24, 69–76, 10.5802/crchim.72.