Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Keiyo Takubo and Version 2 by Catherine Yang.
Hematopoietic stem cells (HSCs) in bone marrow continuously supply a large number of blood cells throughout life in collaboration with hematopoietic progenitor cells (HPCs). HSCs and HPCs are thought to regulate and utilize intracellular metabolic programs to obtain metabolites, such as adenosine triphosphate (ATP), which is necessary for various cellular functions. The metabolic programs of tissue stem/progenitor cells and their underlying molecular mechanisms have been elucidated using a variety of metabolic analysis methods.
- stem cell metabolism
- hematopoiesis
- hematopoietic stem cell
- hypoxia
- metabolomics
- stem cell culture
1. HSCs and Their Metabolic Programs Which Maintain Individual Hematopoiesis
As an example of stem cell metabolism research, applying the various metabolic analysis techniques described above, here thwe researchers present the findings on HSC and HPC metabolism. Of the cells that make up our bodies, about two-thirds are blood cells, mainly erythrocytes. Blood cells play a variety of roles, such as oxygen transport, immune response, and hemostasis, and are indispensable for sustaining organismal life [1][32]. Blood cells produced in mammals after birth are derived from HSCs and HPCs that reside in the bone marrow. HSCs are typical tissue stem cells that retain the ability to self-renew and differentiate into all blood cells [2][3][7,33]. After differentiating into multipotent progenitors with a reduced self-renewal capacity, HSCs differentiate into more committed HPCs with limited differentiation capacity, such as myeloid and lymphoid progenitors, and finally give rise to terminally differentiated blood cells. HSCs are also used in HSC transplantation, a curative therapy for malignant hematopoietic tumors, in which the recipient’s bone marrow is reconstructed and the hematopoietic system is replaced by blood cells derived from donor HSCs. In the steady state, HSCs are in a quiescent state of the cell cycle (G0 phase), and, together with multipotent progenitor cells, they contribute to the continuous production of blood cells. When acute stresses such as transplantation, hemorrhage, inflammation, or infection are loaded, HSCs leave the G0 state and actively proliferate to provide HPCs and differentiated blood cells, contributing to the restoration of homeostasis in the hematopoietic system [4][34].
In order for HSCs to function properly both under steady-state conditions and under stress, the molecular mechanisms that define the identity of stem cells, mainly transcription factors, must be activated. In particular, the production and consumption of ATP, the energy currency, and the maintenance of the nuclear genome, epigenetic state, cell membrane, and intracellular organelles by nucleic acids and lipids are essential for various events such as cell survival, division, and differentiation (Figure 1). The elucidation of these metabolic programs in HSCs and the development of HSC manipulation techniques based on these programs are important research trends [5][35].
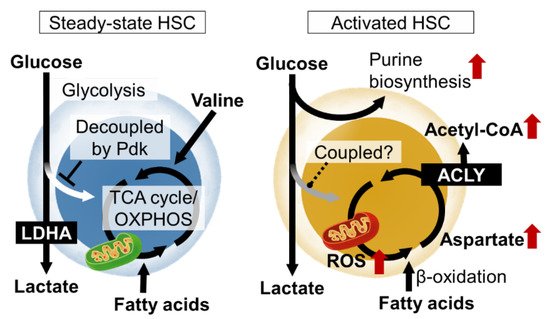
Figure 1. Nutrient requirements and metabolic programs of hematopoietic stem cells (HSCs) in steady state and upon activation. In steady-state HSCs, glycolytic and mitochondrial ATP production are decoupled. Various metabolic pathways are activated in HSCs upon stress loading or cytokine stimulation, resulting in metabolic reprogramming that induces proliferation and differentiation. LDHA, lactate dehydrogenase A; ROS, reactive oxygen species; Pdk, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation; ACLY, ATP citrate lyase.
2. Metabolic Control of HSPCs in Steady State
2.1. Hypoxic Microenvironment and HSCs
Oxygen is required for ATP production in mitochondria. Thus, cellular energy production and oxygen demand are inseparable. While the bone marrow has a high oxygen demand for blood cell production, oxygen supply is dependent on perfusion by blood vessels that penetrate the bone and enter the marrow cavity. In other words, it is difficult to sufficiently and timely increase the oxygen supply through angiogenesis to meet the demand for oxygen and nutrients in the bone marrow. For this reason, bone marrow has long been considered a hypoxic environment. Using the hypoxia probe pimonidazole, HSCs were found to be even more hypoxic than other differentiated blood cells [6][7][8][36,37,38]. Recently, oxygen partial pressure in bone marrow was measured using a combination of in vivo imaging and phosphorescent probes. The analysis reported that the bone marrow is an extremely hypoxic organ with an oxygen partial pressure of approximately 10 mmHg [9][10][39,40]. Indeed, the activation of the hypoxia sensor transcription factor hypoxia-inducible factor-1α (HIF-1α) is observed in HSCs. HSCs lacking HIF-1α were functionally impaired as a result of increased cell cycling, and their stem cell activity decreased [6][36]. This was thought to be due in part to the inability of HSCs to maintain their expression of GRP78, the receptor for Cripto, which maintains stem cell activity [11][41]. In addition, HIF-1α deficiency causes HSCs to leave the bone marrow niche and emerge in the periphery [6][36]. HIF-1α acts as one of the hubs of HSC regulation; for example, HIF-1α reduction due to transcription factor ID2 deficiency leads to HSC activation and depletion [12][42]. Overall, the hypoxic environment in the bone marrow is an environmental factor that could contribute to the maintenance of HSCs.
2.2. Regulation of Glycolysis and Decoupling from TCA Cycle in HSCs
Metabolomic analysis using capillary electrophoresis time-of-flight MS (CE-TOFMS) suggested that the activity of phosphofructokinase (PFK), the rate-limiting enzyme of glycolysis, is high in HSCs, and that the metabolite flux from glycolysis to mitochondria is suppressed [13][23]. HIF-1α is also known as a regulator of glycolysis. In fact, HIF-1α induces the gene expression of various glycolytic enzymes in HSCs. Among the glycolytic enzymes, the deficiency of LDHA, a subunit of the lactate dehydrogenase complex, strongly impairs the number and function of hematopoietic stem cells, suggesting the importance of the glycolytic system for the maintenance of HSCs [14][43]. Interestingly, in mice lacking PKM2, an isozyme of pyruvate kinase and also an enzyme of glycolysis, HSCs are functionally normal, but HPCs are impaired [14][43]. These observations suggest that the metabolic program of HSPCs is maintained at least in part by the expression of specific metabolic enzymes themselves. HIF-1α also induces the expression of pyruvate dehydrogenase kinase (Pdk), which phosphorylate and inactivate pyruvate dehydrogenase (PDH). Among the four Pdk family members, HIF-1α-deficient HSCs show reduced expression of Pdk2 and Pdk4 [13][23]. PDH catalyzes the reaction that converts pyruvate to acetyl CoA, which has the effect of driving the mitochondrial tricarboxylic acid (TCA) cycle. Pdk functions as a negative regulator of the metabolic flux from glycolysis to the mitochondrial TCA cycle. Thus, HIF-1α in HSCs suppresses the metabolic flux into the TCA cycle from glycolysis by Pdk expression. This indicates that ATP production by glycolysis, which does not require oxygen, and mitochondrial ATP production by oxidative phosphorylation (OXPHOS), which does require oxygen, are decoupled. Rather than having the glycolysis and mitochondrial OXPHOS integrated into one ATP-producing system through aerobic glycolysis, the independent existence of the two ATP-producing pathways (anaerobic glycolysis and OXPHOS) may provide stress tolerance for the HSCs. In fact, HSCs lacking these two Pdk family genes showed not only altered metabolic characteristics, such as a decrease in glycolysis and increased mitochondrial mass, but also the activation of cell cycling, accumulation of oxidative stress, and decreased stem cell activity [13][23]. Thus, the metabolic properties of HSCs, which maintain the decoupling of glycolytic and mitochondrial ATP production via the HIF-1α-Pdk axis, are necessary for maintaining the phenotypes and functions of HSCs.
2.3. Mitochondrial Dynamics and Steady-State HSCs
Mitochondrial activity and quantity fission and fusion alter the number and functions of HSCs in a steady state or during stress. Based on analysis using transmission electron microscopy and mitochondrial probes, the number of mitochondria in HSCs was thought to be low. However, it has been reported that HSCs have the ability to pump mitochondrial probes out of the cell and that their actual mitochondrial mass is equal to or rather higher than that of differentiated cells [15][44]. Interestingly, flux analyzer analysis has shown that mitochondrial oxygen consumption is suppressively regulated in HSCs [15][44]. These findings, together with the fact that metabolic flux from glycolysis to the TCA cycle is suppressed under a steady state, suggest that mitochondrial activity may be suppressed in HSCs. However, this does not mean that mitochondria themselves are unnecessary at all. For example, the mitochondrial enzyme PTPMT1 (PTEN-like phosphatase) is required for the maintenance of mitochondria in HSCs and their differentiation potential [16][45]. In addition, the loss of fumarate hydratase (Fh1), an enzyme of the TCA cycle, impairs the self-renewal and differentiation potential of HSCs [17][46]. Furthermore, the loss of Mitofusin 2 (Mfn2), which regulates mitochondrial fusion and division, maintains the ability of HSCs to differentiate into myeloid cells, but impairs differentiation into lymphoid lineages [18][47]. More directly, the loss of the complex III subunit Rieske iron-sulfur protein (RISP) of mitochondrial OXPHOS leads to the proliferation of HSCs along with HPCs, impairing the quiescence, differentiation potential, and survival of fetal and adult HSCs [19][48].
2.4. Environmentally-Derived Metabolites That Maintain HSC Homeostasis
Not only the environment near the HSCs but also the metabolites derived from the diet, are important for the metabolic control of steady-state HSCs. Vitamin A supplied by food activate retinoic acid (RA) signaling functions to maintain quiescence by inhibiting reactive oxygen species production and protein translation in HSCs [20][49]. Importantly, non-classical RA signaling by the Cyp26b1-mediated production of 4-oxo-RA regulates HSC function via RA receptor beta [21][50]. Another example is ascorbate/vitamin C. Metabolomic profiling showed that human and mouse HSCs had unusually high levels of ascorbate, which decreased upon differentiation. The systemic depletion of ascorbate in mice increased HSC frequency and function, in part by the decreased function of Tet2, a dioxygenase tumor suppressor. Ascorbate depletion accelerated leukemogenesis induced by an oncogenic mutant of Flt3. Thus, the accumulation of ascorbate in HSCs increases Tet activity to limit HSC frequency and suppress leukemogenesis [22][51].
3. Metabolic Regulation of HSCs under Stress
3.1. Acute Stress and HSC Metabolism
HSCs are not always quiescent in the cell cycle. Various stresses, or the loss of differentiated blood cells (infection, hemorrhage, transplantation, etc.), activate the HSCs to proliferate symmetrically or asymmetrically and provide differentiated cells. This is a fundamental activity of the HSC to maintain homeostasis in the blood system. Various stress loads activate energy production by mitochondria in HSCs. For example, fatty acid β-oxidation induced by PPARδ reportedly supports HSC self-renewal divisions. Stress-induced mitochondrial activation increases ROS as a byproduct. As a result, the stress sensor p38MAP kinase is activated. HSCs deficient in p38α, the major p38MAP kinase isozyme in blood cells, have impaired hematopoietic recovery with decreased cell cycling after stress loading [23][52]. In the stressed HSCs, p38α initiates CREB-Mitf-inosine monophosphate dehydrogenase 2 (Impdh2) signaling to activate purine metabolism. Impdh2 is the rate-limiting enzyme in the biosynthetic pathway for guanine nucleotide, following the pentose phosphate pathway, a branch pathway of glycolysis. This was found in metabolite profiling via the CE-TOFMS of HSCs after stress loading, as amino acids that should enter the purine biosynthetic pathway accumulate in p38α deficiency. Additionally, HSPCs show a strong dependence on hypoxanthine guanine phosphoribosyl transferase (HPRT)-associated purine salvaging. HPRT deficiency resulted in altered cell-cycle progression, proliferation kinetics, and mitochondrial membrane potential in HSCs, whereas the HPCs were less affected [24][53]. Thus, the activation of purine metabolism is critical for HSC function upon stress. However, it remains unclear how the overall central carbon metabolism is readjusted during stress loading.
3.2. Mitochondrial Activity and Acutely Stressed HSCs
The mitochondrial activation of HSCs by proliferative stress is induced by elevated Ca2+ levels in the cytoplasm [25][54]. This is suppressed under a steady state by the adenosine supplied by the neighboring cells. Based on these results, HSC mitochondria are an important regulator of HSC numbers and function. The electron transport chain in mitochondria supports aspartate production. Aspartate levels in HSCs and HPCs are dependent on intracellular synthesis and increase upon HSC activation. The regulation of aspartate levels is well-coupled with HSC function [26][55]. The overexpression of the glutamate/aspartate transporter, Glast, or the loss of the glutamic-oxaloacetic, transaminase 1 (Got1), each increase the aspartate levels in HSPCs and increase the functioning in HSCs but not HPCs. In contrast, the loss of Got2 reduces aspartate and the functioning in HSCs but not HPCs. The deletion of both Got1 and Got2 eliminated the HSCs. By using isotope tracing, aspartate was identified as a source for the synthesis of asparagine and purines [26][55]. Both contributed to increased HSC function during hematopoietic regeneration. During proliferative stress, metabolism also regulates the epigenetic status of the HSCs. The acetyl CoA supply via ATP citrate lyase supports differentiated cell production via increased histone acetylation in HSCs [27][56].
3.3. Aging Stress and HSC Metabolism
The quality control of mitochondria and other intracellular organelles is important in the metabolic regulation of old stem cells, including HSCs. It is known that mitochondrial accumulation and metabolic activation due to decreased autophagy during aging leads to decreased HSC function [28][31]. Interestingly, one-third of old HSCs still perform autophagy properly, contributing to the maintenance of hematopoiesis through the healthy HSCs after aging [28][31]. Recently, another intracellular organelle quality control mechanism, chaperone-mediated autophagy (CMA), was found to be associated with HSC aging. CMA is required for fatty acid metabolism upon HSC activation and CMA activity in the HSCs decreases with age [29][57]. The restoration of CMA in old HSCs can restore the functionality of old mouse and human HSCs [29][57]. Apart from mitochondrial mass, an appropriate level of mitochondrial membrane potential (MMP) is also an indicator of the quality of HSCs as they age [30][58]. In this study, MMPs in HSCs are measured using TMRM, a fluorescent MMP probe; the addition of TMRM can be reliably measured by an inhibitor of the ABC transporter (verapamil). The pharmacological restoration of MMP improves the function of old HSCs [30][58]. Other metabolic pathways besides mitochondria and HSC aging are still unclear. In HSCs, p38α inhibits aging phenotypes in the early aging stage but promotes aging phenotypes in the late aging stage, and purine metabolism may also undergo a time-dependent change [31][59]. These reports suggest that the reactivation of young HSC-like metabolism may be a promising approach to rejuvenating HSC after aging.
4. Application of Metabolic Findings of HSC
4.1. Metabolic Optimization of HSC Culture
Attempts are also being made to apply the knowledge gained from HSC metabolism research to stem cell culture or manipulation. For example, the treatment of HSCs with the RA signaling agonist all-trans retinoic acid (ATRA) was shown to maintain the immature phenotype, low biosynthetic activity, and cell cycle quiescence [20][49]. ATRA-treated HSCs showed an improved serial plating capacity in vitro and performed with a better serial reconstitution capacity in vivo compared to control cells. Another example is the use of a pyruvate analog, 1-aminoethylphosphinic acid (1-AA), that mimics the action of Pdk. The treatment of HSCs with 1-AA maintained the metabolic program and cellular quiescence of HSCs in vitro in the bone marrow. As a result, HSCs can be cultured for long periods of time while retaining their transplantation activity [13][23].
Recently, metabolites in the bone marrow have been shown to constitute a niche of environmental factors that regulate HSCs. Since adipocyte is known to increase in bone marrow, especially with aging, a close relationship between HSCs and fatty acids in bone marrow was assumed. Outside the cell, fatty acids are mainly bound to albumin. In HSC cultures, only a much lower concentration of albumin (0.1–1%) is added to the culture than the blood concentration of albumin (about 4%). In fact, when HSCs are cultured in vitro in hypoxic conditions with the addition of sufficient amounts of fatty acid-bound albumin, they maintain cell cycle quiescence and transplant survival [32][60]. Although the mode of fatty acid supply from intramedullary fat to the HSCs remains unclear, in vivo imaging and lipidomics analyses suggest that fatty acids abundantly loaded with circulating albumin may diffuse deep into the bone marrow HSC niche to supply the fatty acids.
4.2. Non-Conditional Transplantation with the Removal of Metabolites from the Diet
Another environmentally-derived metabolite for HSCs is valine. By applying MS technology to tissue sections, one amino acid (valine) has been revealed to be abundantly distributed in bone marrow [33][61]. In fact, when amino acids were removed one by one from the culture medium and examined, the removal of valine, in particular, resulted in a failure to maintain the HSCs. Also, mice fed a valine-deficient diet become recipients that can accept HSC transplantation without pretreatment as a result of the removal of the HSC cells from the bone marrow niche [33][61].