Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Weronika Adach and Version 3 by Camila Xu.
Homozygous familial hypercholesterolemia (HoFH) affects an average of 1 in 300,000 subjects. It is a very rare genetic disorder of lipoprotein metabolism. It is caused by mutations in both alleles of the LDL receptor (LDLR) gene and less often by mutations in APOB, the ligand for LDLR and proprotein convertase subtilisin kexin type 9 (PCSK9), a protein that degrades LDLR.
- ANGPTL3 inhibitors
- evinacumab
- familial hypercholesterolemia
- LDL cholesterol
1. Homozygous Familial Hypercholesterolemia
Homozygous familial hypercholesterolemia (HoFH) affects an average of 1 in 300,000 subjects. It is a very rare genetic disorder of lipoprotein metabolism. It is caused by mutations in both alleles of the LDL receptor (LDLR) gene and less often by mutations in APOB, the ligand for LDLR and proprotein convertase subtilisin kexin type 9 (PCSK9), a protein that degrades LDLR [1]. Higher levels of low-density lipoprotein cholesterol (LDL-C) are characterized by genetic changes that show no expression of the LDL receptor (null homozygotes), compared to changes with two non-zero alleles or one zero and one non-null homozygotes, which only partially reduce the LDL receptor activity [2][3][2,3]. These mutations impair the function of the liver to remove LDL-C from the bloodstream, resulting in high total cholesterol and LDL-C [4]. In comparison, LDL particles that bind PCSK9 are targeted for lysosomal degradation and destruction. Loss-of-function mutations of the PCSK9 gene decrease the level of LDL-C and lower the risk of myocardial infarctions in white and black persons and reduce the risk of stroke in black persons. It can be concluded that PCSK9 inhibitors prevent an atherosclerotic cardiovascular event [1].
Patients with HoFH have very high levels of LDL-C from birth, which result in high risks of premature atherosclerosis and other cardiovascular diseases. Clinically, HoFH is characterized by an LDL-C level > 500 mg/dL (>13 mmol/L). Statins and lipid-lowering drugs are largely dependent on the activity of the LDL receptor; therefore, in patients with two null alleles, they may show diminished efficacy. Therefore, most patients with HoFH do not achieve guideline-recommended levels of LDL-C despite treatment with multiple agents [3]. Unfortunately, mutations in patients with familial hypercholesterolemia are associated with an increased probability (up to 3.8 times) of myocardial infarctions under the age of 55 years [5]. Early diagnosis of FH and follow-up, with comprehensive longitudinal care and particular emphasis on aortic valve obstruction and stenosis, are of key importance in the prevention of premature atherosclerotic cardiovascular disease (ASCVD) [2].
According to the 2019 guidelines of the European Society of Cardiology (ESC) and the European Society of Atherosclerosis (EAS), LDL-C levels should be below 1.42 mmol/L (55 mg/dL) in patients at very high risk of ASCVD, below 1.81 mmol/L (70 mg/dL) in patients at high risk, and below 2.59 mmol/L (100 mg/dL) in moderate-risk patients [5].
2. ANGPTL3, 4, and 8 Protein System-Characteristics and Role in Lipid Metabolism
The angiopoietin-like proteins (ANGPTLs) are a family of proteins consisting of members 1–8 of the angiopoietins, which differ in terms of tissue expression and regulation. They each consist of a common domain at the amino terminus (N-terminal), a coiled-coil domain (CCD), a fibrinogen-like domain (FLD) at the C-terminus of the carboxyl, and a linker region. Angiopoietin-8 differs from the other ANGPTLs in that it does not contain a fibrinogen-like domain at the C-terminus [6]. ANGPTL proteins belong to the vascular endothelial growth factor (VEGF) family and play various roles in biological and pathological processes, including hormone regulation, glucose metabolism, and insulin resistance [7].
ANGPTL3, ANGPTL4, and ANGPTL8 are most important in lipoprotein metabolism because they are responsible for the metabolism of triglycerides (TGs)—rich lipoproteins (chylomicrons, VLDL)—by inhibiting the activities of lipoprotein lipase (LPL), VLDL, and LDL mediated by the inhibition of endothelial lipase (EL) [6][8][6,8]. LPL activity is reduced by changing the conformation from homodimeric, which is biologically active, to biologically inactive, or monomeric. LPL is an enzyme produced in fat and muscle cells that limits the rate of hydrolysis of TG-rich lipoproteins to free fatty acids (FFA). When this process is disturbed, severe hypertriglyceridemia occurs in plasma [9]. The best-known ANGPTL is ANGPTL3, which was discovered in 1999. ANGPTL3 is produced in the liver. In the following year, 2000, ANGPTL4 was discovered, and it is produced in the liver, skeletal muscle, adipose tissue, gut, brain, and heart. Additionally, ANGPTL8 was discovered in 2012, and its main source is adipose tissue and the liver [10].
ANGPTL3, 4, and 8 control the availability of triglyceride-rich lipoproteins, LDL, and high-density lipoprotein cholesterol (HDL-C), depending on the nutritional status of the body, temperature, and physical activity, by regulating LPL secretion. LPL activity is increased after a meal, and triglycerides are stored in the white adipose tissue of WAT. In contrast, after a meal, LPL activity is reduced in the heart, brown adipose tissue, and skeletal muscle by ANGPTL3 and 8 (ANGPTL8 expression is especially increased). The opposite occurs during fasting, where LPL activity increases in the heart, brown adipose tissue, and skeletal muscle. In white adipose tissue, the activity of LPL during fasting is reduced by ANGPTL4 [11][12][13][14][15][11,12,13,14,15] (Figure 1).
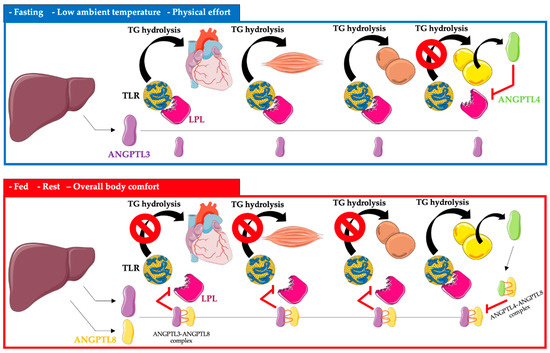
FRycigurena 1. Regulation of triglyceride metabolism in heart, muscle, brown adipose tissue, and white adipose tissue by ANGPTL3, ANGPTL4, and ANGPTL8. Abbreviations: TG triglyceride; TLR—triglyceride-rich lipoprotein; LPL—lipoprotein lipase; ANGPTL3—angiopoietin-like protein 3; ANGPTL4—angiopoietin-like protein 4; ANGPTL8—angiopoietin-like protein 8. The following was used in the preparation of the figure: https://smart.servier.com (free-access; 20 October 2022).
ANGPTL3, apart from its effect on LPL, also reduces the activity of EL, which leads to a slowdown in the metabolism of triglyceride-rich lipoproteins [16].
3. ANGPTL3, 4, and 8 as Biomarkers of Cardiovascular Risk
Several publications have reported that ANGPTL3 deficiency protects against coronary artery disease (CAD). According to the research of Stitziel et al., in subjects with complete ANGPTL3 deficiency, the coronary arteries lacked atherosclerotic plaque [12]. Moreover, healthy patients showed lower concentrations of ANGPTL3 compared to patients who experienced myocardial infarctions (MIs) [12]. In patients with ANGPTL33 concentrations of 18–271 ng/mL, the risk of a heart attack was reduced by up to 29%. Researchers also demonstrated an association of the loss-of-function (LOF) mutation in ANGPTL3 with the risk of CAD. The levels of low-density lipoprotein LDL-C, high-density HDL-C, and TGs are dependent on the LOF in ANGPTL3. Patients carrying the LOF mutation showed a 34% reduction in the risk of CAD compared to patients who did not carry the LOF mutation. In addition, patients with the LOF mutation showed 11% lower total cholesterol, 12% lower LDL, and 17% lower TG levels compared to those without the mutation. In addition to the fact that the loss of ANGPTL3 increases LPL activity, leading to a reduction in TGs and LDL-rich lipoproteins, it may affect insulin sensitivity and play an important role in glucose hemostasis [12].
In another study on the effects of ANGPTL3 and 4 on CAD, the team of Sun et al. presented the results of a study involving 305 patients. A high level of ANGPTL3 was closely related to the severity of atherosclerotic lesions in the coronary vessels, while the level of ANGPTL4 was reduced. The levels of these glycoproteins may have significant impacts on the development of CAD [17]. Another study showed the relationship between mutations inactivating the ANGPTL4 gene on the risk of ischemic heart disease. This researchtudy included over 42,000 subjects. Dewey et al. in 2017 proved that the reductions in the levels of TGs, total cholesterol, and LDL-C were caused by the inactivation of ANGPTL4 through the heterozygous mutation E40k. Patients with this ANGPTL4 mutation showed a 19% lower risk of coronary heart disease [18]. In a similar study led by Stitziel et al., patients with the E40K ANGPTL4 mutation showed about 35% lower TG concentration. Additionally, the risk of coronary heart disease was 53% lower. However, no significant effect of ANGPTL4 p.E40K on LDL-C was observed [19]. The research team of Gusarova et al. showed the effect of the E40K ANGPTL4 mutation on the reduction of the risk of type-2 diabetes by 12%. This researchtudy was conducted on 58,000 participants in the DiscovEHR Study [20]. Similar results were presented by the team of Klarin et al. in a study of 310,000 subjects. The effect of the loss-of-function (LOF) ANGPTL4 mutation on the risk of ischemic heart disease and type-2 diabetes was assessed. It was shown that the risk of ischemic heart disease was reduced by 16% and the risk of type 2 diabetes was reduced by 12% [21].
presented in Figure Figure 22.
4. Evinacumab-Structure and the Mechanism of Action
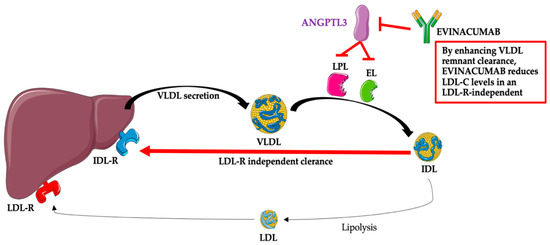
Evinacumab (Evkeeza®; formerly RENG1500) is a fully human monoclonal antibody, inhibiting circulating ANGPTL3, which was invented by Regeneron Pharmaceuticals Inc. [3] and manufactured with the use of the cell culture method with genetically engineered recombinant Chinese hamster ovary cells [22]. Evinacumab is an IgG4 monoclonal antibody consisting of two disulfide-linked human heavy chains (453 amino acids each) and human kappa light chains (214 amino acids). Heavy chains are covalently linked by disulfide bonds to light chains [22]. Evinacumab was approved by the US Food and Drug Administration on February 2021 and the European Medicines Agency (EMA) in June 2021 and is now available on the market under the trade name Evkeeza to treat adult and adolescent patients (≥12 years) with homozygous familial hypercholesterolemia [23]. The recommended dose of this new drug is 15 mg/kg, administered by intravenous infusion (IV) over one hour once monthly [23]. After administration, evinacumab binds its target, ANGPTL3, and inhibits its function, leading to increased LPL and EL activities and lower TG, LDL-C, and HDL-C plasma levels [24]. The mechanism associated with the reduction of LDL-C by evinacumab is not fully known; however, this effect is independent of the LDL receptor and, thus, probably due to the promotion of very-low-density lipoprotein (VLDL) processing and the upstream clearance of LDL formation [12][13][14][15][24][12,13,14,15,24]. The mechanism of the action of evinacumab ispresented in Figure Figure 22.
Figure 2. Evinacumab—mechanism of action. Abbreviations: ANGPTL3—angiopoietin-like protein 3; EL—endothelial lipase; IDL—intermediate-density lipoprotein (VLDL remnants); IDL-R—intermediate-density lipoprotein receptor (VLDL remnant receptor); LDL—low-density lipoprotein; LDL-C—low-density lipoprotein; LDL-R—low-density lipoprotein receptor; LPL—lipoprotein lipase; VLDL—very-low-density lipoprotein. The following was used in the preparation of the figure: https://smart.servier.com (free-access; 20 October 2022).