Coronary heart disease (CHD) is the leading cause of death around the world. Based on the roles of vascular endothelial growth factor (VEGF) family members to regulate blood and lymphatic vessels and metabolic functions, several therapeutic approaches have been attempted during the last decade. However proangiogenic therapies based on classical VEGF-A have been disappointing. Therefore, it has become important to focus on other VEGFs, like VEGF-B, which is a novel member of the VEGF family. Recent studies have shown very promising potential of the VEGF-B to treat CHD and heart failure. This review article aims to present the role of VEGF-B in endothelial biology and as a potential therapeutic agent for CHD and heart failure.
- angiogenesis
- CHD
- gene therapy
- VEGF-B
1. Vascular Endothelial Growth Factor B
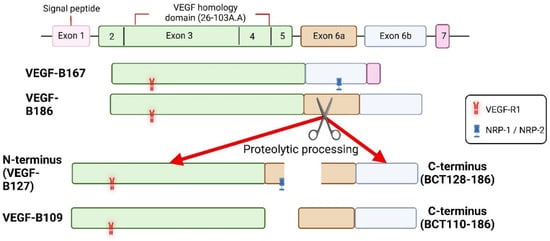
2. Cardioprotective Role of VEGF-B
2.1. Unique Neovascularization in the Heart
2.2. Anti-Apoptosis
2.3. Antioxidant Activity
2.4. Distinct Metabolic Reprogramming
2.5. Cardiac Hypertrophy
2.6. Cardiac Contractility
3. Summary
In addition to its angiogenic role, the regulation of myocardial contractility and metabolism as well as the cardioprotective role of VEGF-B have made it distinct from the other pro-angiogenic factors of the VEGF family (Figure 2) [62]. Cumulatively, the above data suggest that VEGF-B is a strong candidate for the treatment of CHD and heart failure.
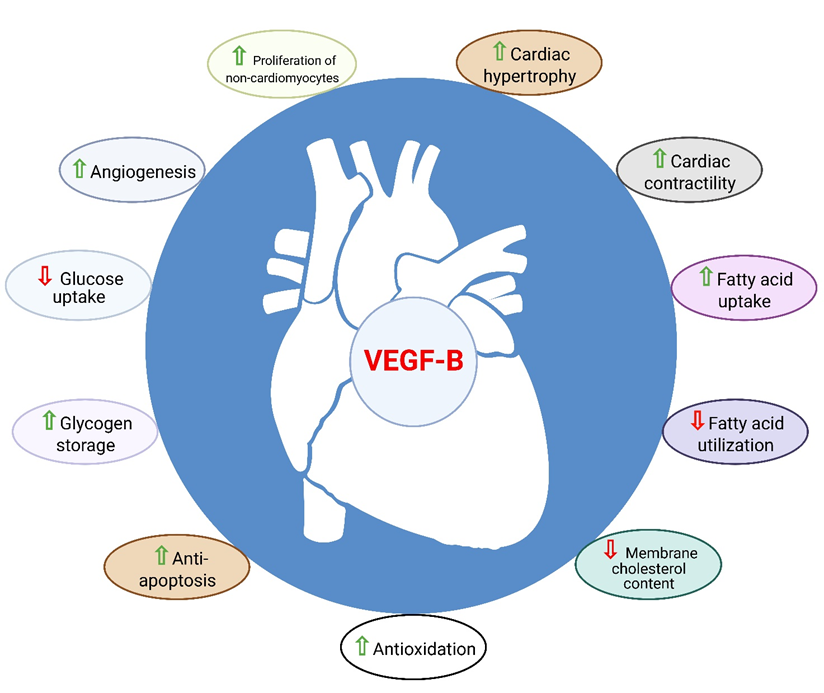
Figure 2. Role of VEGF-B in the heart. VEGF-B is known to have cardioprotective properties such as angio-genesis, anti-apoptosis, anti-oxidation, metabolic reprograming, cardiac contractility and physio-logical cardiac hypertrophy.
References
- Olofsson, B.; Korpelainen, E.; Pepper, M.S.; Mandriota, S.J.; Aase, K.; Kumar, V.; Gunji, Y.; Jeltsch, M.M.; Shibuya, M.; Alitalo, K.; et al. Vascular Endothelial Growth Factor B (VEGF-B) Binds to VEGF Receptor-1 and Regulates Plasminogen Activator Activity in Endothelial Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11709–11714.
- Aase, K.; von Euler, G.; Li, X.; Pontén, A.; Thorén, P.; Cao, R.; Cao, Y.; Olofsson, B.; Gebre-Medhin, S.; Pekny, M.; et al. Vascular Endothelial Growth Factor-B-Deficient Mice Display an Atrial Conduction Defect. Circulation 2001, 104, 358–364.
- Hagberg, C.E.; Mehlem, A.; Falkevall, A.; Muhl, L.; Fam, B.C.; Ortsäter, H.; Scotney, P.; Nyqvist, D.; Samén, E.; Lu, L.; et al. Targeting VEGF-B as a Novel Treatment for Insulin Resistance and Type 2 Diabetes. Nature 2012, 490, 426–430.
- Lv, Y.X.; Zhong, S.; Tang, H.; Luo, B.; Chen, S.J.; Chen, L.; Zheng, F.; Zhang, L.; Wang, L.; Li, X.Y.; et al. VEGF-A and VEGF-B Coordinate the Arteriogenesis to Repair the Infarcted Heart with Vagus Nerve Stimulation. Cell. Physiol. Biochem. 2018, 48, 433–449.
- Feng, L.; Ren, J.; Li, Y.; Yang, G.; Kang, L.; Zhang, S.; Ma, C.; Li, J.; Liu, J.; Yang, L.; et al. Resveratrol Protects against Isoproterenol Induced Myocardial Infarction in Rats through VEGF-B/AMPK/ENOS/NO Signalling Pathway. Free Radic. Res. 2019, 53, 82–93.
- Enholm, B.; Paavonen, K.; Ristimäki, A.; Kumar, V.; Gunji, Y.; Klefstrom, J.; Kivinen, L.; Laiho, M.; Olofsson, B.; Joukov, V.; et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 MRNA Regulation by Serum, Growth Factors, Oncoproteins and Hypoxia. Oncogene 1997, 14, 2475–2483.
- Monastero, R.; García-Serrano, S.; Lago-Sampedro, A.; Rodríguez-Pacheco, F.; Colomo, N.; Morcillo, S.; Martín-Nuñez, G.M.; Gomez-Zumaquero, J.M.; García-Fuentes, E.; Soriguer, F.; et al. Methylation Patterns of Vegfb Promoter Are Associated with Gene and Protein Expression Levels: The Effects of Dietary Fatty Acids. Eur. J. Nutr. 2017, 56, 715–726.
- Lal, N.; Puri, K.; Rodrigues, B. Vascular Endothelial Growth Factor B and Its Signaling. Front. Cardiovasc. Med. 2018, 5, 39.
- Li, X.; Aase, K.; Li, H.; von Euler, G.; Eriksson, U. Isoform-Specific Expression of VEGF-B in Normal Tissues and Tumors. Growth Factors 2001, 19, 49–59.
- Li, X. VEGF-B: A Thing of Beauty. Cell Res. 2010, 20, 741–744.
- Aase, K.; Lymboussaki, A.; Kaipainen, A.; Olofsson, B.; Alitalo, K.; Eriksson, U. Localization of VEGF-B in the Mouse Embryo Suggests a Paracrine Role of the Growth Factor in the Developing Vasculature. Dev. Dyn. 1999, 215, 12–25.
- Shibuya, M. Structure and Dual Function of Vascular Endothelial Growth Factor Receptor-1 (Flt-1). Int. J. Biochem. Cell Biol. 2001, 33, 409–420.
- Mallick, R.; Gurzeler, E.; Toivanen, P.I.; Nieminen, T.; Ylä-Herttuala, S. Novel Designed Proteolytically Resistant VEGF-B186R127S Promotes Angiogenesis in Mouse Heart by Recruiting Endothelial Progenitor Cells. Front. Bioeng. Biotechnol. 2022, 10, 907538.
- Eldrid, C.; Zloh, M.; Fotinou, C.; Yelland, T.; Yu, L.; Mota, F.; Selwood, D.L.; Djordjevic, S. VEGFA, B, C: Implications of the C-Terminal Sequence Variations for the Interaction with Neuropilins. Biomolecules 2022, 12, 372.
- Bry, M.; Kivelä, R.; Leppänen, V.M.; Alitalo, K. Vascular Endothelial Growth Factor-B in Physiology and Disease. Physiol. Rev. 2014, 94, 779–794.
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264.
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular Endothelial Growth Factor B Controls Endothelial Fatty Acid Uptake. Nature 2010, 464, 917–921.
- Nash, A.D.; Baca, M.; Wright, C.; Scotney, P.D. The Biology of Vascular Endothelial Growth Factor-B (VEGF-B). Pulm. Pharmacol. Ther. 2006, 19, 61–69.
- Lagercrantz, J.; Farnebo, F.; Larsson, C.; Tvrdik, T.; Weber, G.; Piehl, F. A Comparative Study of the Expression Patterns for Vegf, Vegf-b/Vrf and Vegf-c in the Developing and Adult Mouse. Biochim. Biophys. Acta Gene Struct. Expr. 1998, 1398, 157–163.
- Bellomo, D.; Headrick, J.P.; Silins, G.U.; Paterson, C.A.; Thomas, P.S.; Gartside, M.; Mould, A.; Cahill, M.M.; Tonks, I.D.; Grimmond, S.M.; et al. Mice Lacking the Vascular Endothelial Growth Factor-B Gene (Vegfb) Have Smaller Hearts, Dysfunctional Coronary Vasculature, and Impaired Recovery from Cardiac Ischemia. Circ. Res. 2000, 86, e29–e35.
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419.
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A Single–Cell Type Transcriptomics Map of Human Tissues. Sci. Adv. 2021, 7, eabh2169.
- Muhl, L.; Moessinger, C.; Adzemovic, M.Z.; Dijkstra, M.H.; Nilsson, I.; Zeitelhofer, M.; Hagberg, C.E.; Huusko, J.; Falkevall, A.; Ylä-Herttuala, S.; et al. Expression of Vascular Endothelial Growth Factor (VEGF)-B and Its Receptor (VEGFR1) in Murine Heart, Lung and Kidney. Cell Tissue Res. 2016, 365, 51–63.
- Kivelä, R.; Bry, M.; Robciuc, M.R.; Räsänen, M.; Taavitsainen, M.; Silvola, J.M.; Saraste, A.; Hulmi, J.J.; Anisimov, A.; Mäyränpää, M.I.; et al. VEGF-B-Induced Vascular Growth Leads to Metabolic Reprogramming and Ischemia Resistance in the Heart. EMBO Mol. Med. 2014, 6, 307–321.
- Lal, N.; Chiu, A.P.L.; Wang, F.; Zhang, D.; Jia, J.; Wan, A.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Loss of VEGFB and Its Signaling in the Diabetic Heart Is Associated with Increased Cell Death Signaling. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1163–H1175.
- Devaux, Y.; Vausort, M.; Azuaje, F.; Vaillant, M.; Lair, M.L.; Gayat, E.; Lassus, J.; Ng, L.L.; Kelly, D.; Wagner, D.R.; et al. Low Levels of Vascular Endothelial Growth Factor B Predict Left Ventricular Remodeling after Acute Myocardial Infarction. J. Card. Fail. 2012, 18, 330–337.
- Karpanen, T.; Bry, M.; Ollila, H.M.; Seppänen-Laakso, T.; Liimatta, E.; Leskinen, H.; Kivelä, R.; Helkamaa, T.; Merentie, M.; Jeltsch, M.; et al. Overexpression of Vascular Endothelial Growth Factor-B in Mouse Heart Alters Cardiac Lipid Metabolism and Induces Myocardial Hypertrophy. Circ. Res. 2008, 103, 1018–1026.
- Lähteenvuo, J.E.; Lähteenvuo, M.T.; Kivelä, A.; Rosenlew, C.; Falkevall, A.; Klar, J.; Heikura, T.; Rissanen, T.T.; Vähäkangas, E.; Korpisalo, P.; et al. Vascular Endothelial Growth Factor-B Induces Myocardium-Specific Angiogenesis and Arteriogenesis via Vascular Endothelial Growth Factor Receptor-1- and Neuropilin Receptor-1-Dependent Mechanisms. Circulation 2009, 119, 845–856.
- Huusko, J.; Lottonen, L.; Merentie, M.; Gurzeler, E.; Anisimov, A.; Miyanohara, A.; Alitalo, K.; Tavi, P.; Ylä-Herttuala, S. AAV9-Mediated VEGF-B Gene Transfer Improves Systolic Function in Progressive Left Ventricular Hypertrophy. Mol. Ther. 2012, 20, 2212–2221.
- Räsänen, M.; Sultan, I.; Paech, J.; Hemanthakumar, K.A.; Yu, W.; He, L.; Tang, J.; Sun, Y.; Hlushchuk, R.; Huan, X.; et al. VEGF-B Promotes Endocardium-Derived Coronary Vessel Development and Cardiac Regeneration. Circulation 2021, 143, 65–77.
- Korpela, H.; Hätinen, O.-P.; Nieminen, T.; Mallick, R.; Toivanen, P.; Airaksinen, J.; Valli, K.; Hakulinen, M.; Poutiainen, P.; Nurro, J.; et al. Adenoviral VEGF-B186R127S Gene Transfer Induces Angiogenesis and Improves Perfusion in Ischemic Heart. iScience 2021, 24, 103533.
- Nurro, J.; Halonen, P.J.; Kuivanen, A.; Tarkia, M.; Saraste, A.; Honkonen, K.; Lähteenvuo, J.; Rissanen, T.T.; Knuuti, J.; Ylä-Herttuala, S. AdVEGF-B186 and AdVEGF-DΔNΔC Induce Angiogenesis and Increase Perfusion in Porcine Myocardium. Heart 2016, 102, 1716–1720.
- Robciuc, M.R.; Kivelä, R.; Williams, I.M.; de Boer, J.F.; van Dijk, T.H.; Elamaa, H.; Tigistu-Sahle, F.; Molotkov, D.; Leppänen, V.M.; Käkelä, R.; et al. VEGFB/VEGFR1-Induced Expansion of Adipose Vasculature Counteracts Obesity and Related Metabolic Complications. Cell Metab. 2016, 23, 712–724.
- Li, Y.; Zhang, F.; Nagai, N.; Tang, Z.; Zhang, S.; Scotney, P.; Lennartsson, J.; Zhu, C.; Qu, Y.; Fang, C.; et al. VEGF-B Inhibits Apoptosis via VEGFR-1-Mediated Suppression of the Expression of BH3-Only Protein Genes in Mice and Rats. J. Clin. Investig. 2008, 118, 913–923.
- Rowe, G.C.; Young, M.E. VEGF-B: Friend or Foe to the Heart in Times of Nutrient Excess? Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H244–H247.
- Arjunan, P.; Lin, X.; Tang, Z.; Du, Y.; Kumar, A.; Liu, L.; Yin, X.; Huang, L.; Chen, W.; Chen, Q.; et al. VEGF-B Is a Potent Antioxidant. Proc. Natl. Acad. Sci. USA 2018, 115, 10351–10356.
- Shang, R.; Lal, N.; Lee, C.S.; Zhai, Y.; Puri, K.; Seira, O.; Boushel, R.C.; Sultan, I.; Räsänen, M.; Alitalo, K.; et al. Cardiac-Specific VEGFB Overexpression Reduces Lipoprotein Lipase Activity and Improves Insulin Action in Rat Heart. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E753–E765.
- Moessinger, C.; Nilsson, I.; Muhl, L.; Zeitelhofer, M.; Heller Sahlgren, B.; Skogsberg, J.; Eriksson, U. VEGF-B Signaling Impairs Endothelial Glucose Transcytosis by Decreasing Membrane Cholesterol Content. EMBO Rep. 2020, 21, e49343.
- Mallick, R.; Basak, S.; Duttaroy, A.K. Fatty Acids and Evolving Roles of Their Proteins in Neurological, Cardiovascular Disorders and Cancers. Prog. Lipid Res. 2021, 83, 101116.
- Bugger, H.; Abel, E.D. Molecular Mechanisms of Diabetic Cardiomyopathy. Diabetologia 2014, 57, 660–671.
- Randle, P.J.; Hales, C.N.; Garland, P.B.; Newsholme., E.A. The Glucose Fatty-Acid Cycle—Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 281, 785–789.
- Pascual, F.; Coleman, R.A. Fuel Availability and Fate in Cardiac Metabolism: A Tale of Two Substrates. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1425–1433.
- Kim, M.S.; Wang, Y.; Rodrigues, B. Lipoprotein Lipase Mediated Fatty Acid Delivery and Its Impact in Diabetic Cardiomyopathy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 800–808.
- Chou, E.; Suzuma, I.; Way, K.J.; Opland, D.; Clermont, A.C.; Naruse, K.; Suzuma, K.; Bowling, N.L.; Vlahos, C.J.; Aiello, L.P.; et al. Decreased Cardiac Expression of Vascular Endothelial Growth Factor and Its Receptors in Insulin-Resistant and Diabetic States: A Possible Explanation for Impaired Collateral Formation in Cardiac Tissue. Circulation 2002, 105, 373–379.
- Stinkens, R.; Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Targeting Fatty Acid Metabolism to Improve Glucose Metabolism. Obes. Rev. 2015, 16, 715–757.
- Mallick, R.; Duttaroy, A.K. Modulation of Endothelium Function by Fatty Acids. Mol. Cell. Biochem. 2022, 477, 15–38.
- Zhi, Y.F.; Prins, J.B.; Marwick, T.H. Diabetic Cardiomyopathy: Evidence, Mechanisms, and Therapeutic Implications. Endocr. Rev. 2004, 25, 543–567.
- An, D.; Rodrigues, B. Role of Changes in Cardiac Metabolism in Development of Diabetic Cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1489–H1506.
- Dijkstra, M.H.; Pirinen, E.; Huusko, J.; Kivelä, R.; Schenkwein, D.; Alitalo, K.; Ylä-Herttuala, S. Lack of Cardiac and High-Fat Diet Induced Metabolic Phenotypes in Two Independent Strains of Vegf-b Knockout Mice. Sci. Rep. 2014, 4, 6238.
- Yang, Q. Carnitine Palmitoyltransferase 1b Deficiency Protects Mice from Diet-Induced Insulin Resistance. J. Diabetes Metab. 2014, 5, 361.
- Kim, T. Carnitine Palmitoyltransferase 1b Deficient Mice Develop Severe Insulin Resistance after Prolonged High Fat Diet Feeding. J. Diabetes Metab. 2014, 5, 1000401.
- Tirronen, A.; Huusko, J.; Lehtonen, M.; Hamalainen, W.; Hokkanen, K.; Auriola, S.; Yla-Herttuala, S. Overexpression of VEGF-B Alters Cardiac Lipid Metabolism and Predisposes to Heart Failure. Eur. Heart J. 2022, 43, ehac544-753.
- Huusko, J.; Merentie, M.; Dijkstra, M.H.; Ryhänen, M.M.; Karvinen, H.; Rissanen, T.T.; Vanwildemeersch, M.; Hedman, M.; Lipponen, J.; Heinonen, S.E.; et al. The Effects of VEGF-R1 and VEGF-R2 Ligands on Angiogenic Responses and Left Ventricular Function in Mice. Cardiovasc. Res. 2010, 86, 122–130.
- Zentilin, L.; Puligadda, U.; Lionetti, V.; Zacchigna, S.; Collesi, C.; Pattarini, L.; Ruozi, G.; Camporesi, S.; Sinagra, G.; Pepe, M.; et al. Cardiomyocyte VEGFR-1 Activation by VEGF-B Induces Compensatory Hypertrophy and Preserves Cardiac Function after Myocardial Infarction. FASEB J. 2010, 24, 1467–1478.
- Bry, M.; Kivelä, R.; Holopainen, T.; Anisimov, A.; Tammela, T.; Soronen, J.; Silvola, J.; Saraste, A.; Jeltsch, M.; Korpisalo, P.; et al. Vascular Endothelial Growth Factor-B Acts as a Coronary Growth Factor in Transgenic Rats without Inducing Angiogenesis, Vascular Leak, or Inflammation. Circulation 2010, 122, 1725–1733.
- Fong, G.H.; Klingensmith, J.; Wood, C.R.; Rossant, J.; Breitman, M.L. Regulation of Flt-1 Expression during Mouse Embryogenesis Suggests a Role in the Establishment of Vascular Endothelium. Dev. Dyn. 1996, 207, 1–10.
- Unger, R.H. Minireview: Weapons of Lean Body Mass Destruction: The Role of Ectopic Lipids in the Metabolic Syndrome. Endocrinology 2003, 144, 5159–5165.
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A Novel Mouse Model of Lipotoxic Cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822.
- Naumenko, N.; Huusko, J.; Tuomainen, T.; Koivumäki, J.T.; Merentie, M.; Gurzeler, E.; Alitalo, K.; Kivelä, R.; Ylä-Herttuala, S.; Tavi, P. Vascular Endothelial Growth Factor-B Induces a Distinct Electrophysiological Phenotype in Mouse Heart. Front. Physiol. 2017, 8, 373.
- Lähteenvuo, J.; Hätinen, O.P.; Kuivanen, A.; Huusko, J.; Paananen, J.; Lähteenvuo, M.; Nurro, J.; Hedman, M.; Hartikainen, J.; Laham-Karam, N.; et al. Susceptibility to Cardiac Arrhythmias and Sympathetic Nerve Growth in VEGF-B Overexpressing Myocardium. Mol. Ther. 2020, 28, 1731–1740.
- Tirronen, A.; Downes, N.L.; Huusko, J.; Laakkonen, J.P.; Tuomainen, T.; Tavi, P.; Hedman, M.; Ylä-herttuala, S. The Ablation of Vegfr-1 Signaling Promotes Pressure Overload-induced Cardiac Dysfunction and Sudden Death. Biomolecules 2021, 11, 452.
- Ylä-Herttuala, S.; Bridges, C.; Katz, M.G.; Korpisalo, P. Angiogenic Gene Therapy in Cardiovascular Diseases: Dream or Vision? Eur. Heart J. 2017, 38, 1365–1371.