Metabolically active tissues including the heart muscle depend on fatty acids as the predominant source of energy
[42][64]. In diabetes, the heart cannot effectively utilize glucose as an energy source and relies mostly on fatty acids for energy production
[43][65]. Energy production through fatty acid oxidation requires more oxygen than glucose utilization and in diabetes, VEGF-A mediated angiogenesis is obscured due to the reduced oxygen supply
[44][45][66,67]. Concerning insulin resistance, imbalance of fatty acid uptake and oxidation due to the lack of oxygen results in the accumulation of ceramides and diacylglycerols, which amends the effects of insulin mediated glucose uptake related effects on gene expression and signaling in the heart
[40][46][47][48][1,62,68,69]. As VEGF-B is able to induce ceramide accumulation and mitochondrial dysfunction in the heart due to the imbalance of fatty acid uptake and oxidation
[27][42], the therapeutic use of VEGF-B to treat heart failure in diabetes or dyslipidemia could also have disadvantages. However, it is noteworthy to mention that the fatty acid oxidation-related effects of VEGF-B on glucose metabolism were all reported from transgenic and knockout mice on a high-fat diet and were not always repeatable
[3][17][27][49][18,32,42,70]. Thus, the possibility of a high-fat diet induced insulin resistance cannot be excluded
[50][51][71,72]. Corroborating these findings, Kivelä et al. demonstrated the shift of fatty acid oxidation toward glucose metabolism by VEGF-B in the heart of transgenic rats
[24][39]. Similarly, viral-vector mediated VEGF-B186 gene transfer into mouse adipose tissue supports beneficial outcomes such as insulin sensitivity, glucose tolerance, and improved metabolic health
[33][56]. In addition, Tirronen et al. found reduced lipid deposition in the heart, but predisposition to heart failure by VEGF-B in transgenic mice
[52][73]. Combining all the information, it can be suggested that due to the transient duration of the adenoviral vector-based VEGF-B186 gene therapy, this treatment might still be beneficial in heart failure patients with dyslipidemia.
2.5. Cardiac Hypertrophy
VEGF-B mediated cardiac hypertrophy is thought to be dependent on VEGF-R1 expression in cardiomyocytes
[53][54][55][56][46,50,51,74]. Unlike PlGF, VEGF-B mediated cardiac hypertrophy is independent of nitric oxide mediated mechanism as well as NRP-1 mediated signaling
[24][55][39,51]. It is noteworthy to mention that the expression of cytoplasmic bone marrow kinase in chromosome X in arterial endothelium is crucial for VEGF-B induced cardiac hypertrophy
[55][51]. However, the possibility of the involvement of paracrine signaling between cardiomyocytes and endothelial cells cannot be excluded
[28][55][57][58][43,51,75,76]. Involvement of cell types other than cardiomyocytes in the VEGF-B mediated cardiac hypertrophy has been suggested as α-smooth muscle actin-positive cells and endothelial cells show clear proliferative activity in the VEGF-B transgene treated heart
[13][28][28,43].
2.6. Cardiac Contractility
It has been reported that VEGF-B can maintain cardiac contractility
[24][39]. VEGF-B has been found to preserve cardiac function and to induce physiological hypertrophy. VEGF-B167 induces
α-MHC,
SERCA2a,
RYR,
ANF,
BNP, and
PGC1α genes and represses the expression of
β-MHC and
α-actin 1 genes, which attenuate the loss of myocardial mass and maintain cardiac contractility
[54][50]. Additionally, VEGF-B overexpression was found to have effects on cardiac electrophysiology (e.g., downregulation of sodium current (I
Na), transient outward current (I
to), and total K
+ current (I
peak)) as well as hinder Ca
2+ signaling in the mouse heart
[59][77]. VEGF-B186 transgene delivery has been shown to improve cardiac ejection fraction following myocardial infarction
[30][52]. Recently, VEGF-B overexpressed mice (under dobutamine stress) as well as VEGF-B186 and soluble VEGF-R1 transgene co-delivery in pigs were shown to induce cardiac arrhythmias, suggesting the role of NRP-1 in the growth of sympathetic nerves and subsequent cardiac arrhythmias
[60][54]. However, recent findings in mice and porcine studies using mutant unsplicable VEGF-B186 did not support the role of NRP-1 in provoking cardiac arrhythmias
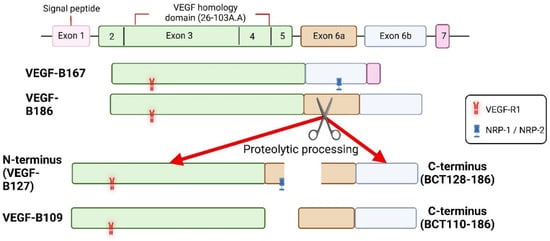
[13][30][31][28,45,52]. Instead, Korpela et al. reported that the C-terminal fragment of the proteolytically spliced VEGF-B186 induced cardiac arrhythmias
[31][45]. As VEGF-R1 is known to regulate cardiac performance
[61][78], it is possible that due to the inability to bind to the VEGF-R1, the C-terminal end of the VEGF-B186 provokes arrhythmias via binding to an unidentified receptor in the heart.
3. Summary
In addition to its angiogenic role, the regulation of myocardial contractility and metabolism as well as the cardioprotective role of VEGF-B have made it distinct from the other pro-angiogenic factors of the VEGF family (Figure 2) [62]. Cumulatively, the above data suggest that VEGF-B is a strong candidate for the treatment of CHD and heart failure.
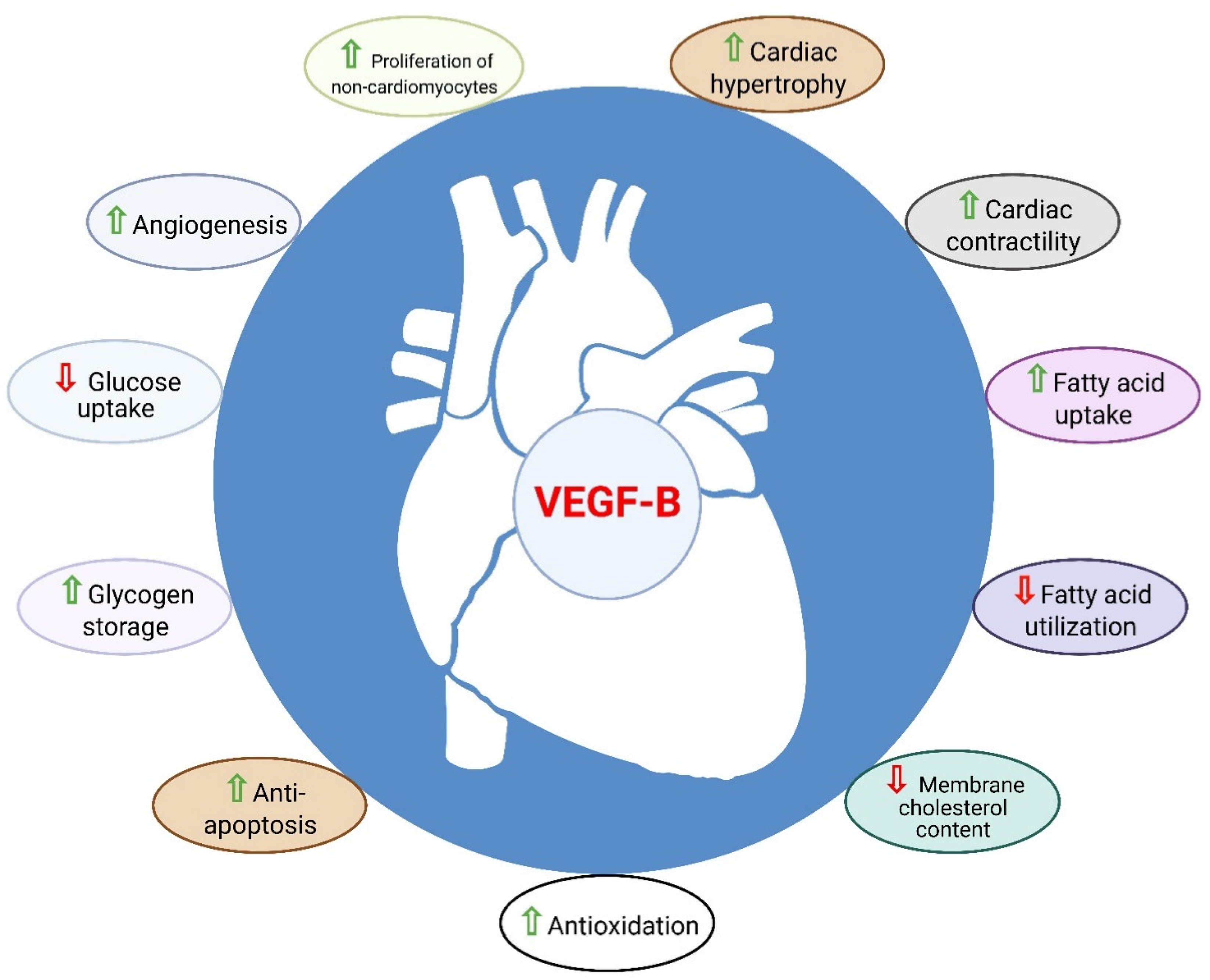
Figure 2. Role of VEGF-B in the heart. VEGF-B is known to have cardioprotective properties such as angio-genesis, anti-apoptosis, anti-oxidation, metabolic reprograming, cardiac contractility and physio-logical cardiac hypertrophy.