Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Irena Baranowska-Bosiacka and Version 2 by Sirius Huang.
CXC motif chemokine ligand 1 (CXCL1), a cytokine belonging to the CXC sub-family of chemokines with CXC motif chemokine receptor 2 (CXCR2) as its main receptor, causes the migration and infiltration of neutrophils to the sites of high expression. This implicates CXCL1 in many adverse conditions associated with inflammation and the accumulation of neutrophils. Herein, the significance of CXCL1 in selected diseases of the cardiovascular system is described.
- CXCL1
- CXCR2
- atherosclerosis
- cardiovascular system
1. The Physiology of Blood Vessels
CXCL1 is important in blood vessel function due to its pro-angiogenic properties–promoting the CXCR2-dependent proliferation, migration and tube formation of endothelial cells [1][2][31,32]. CXCL1 expression is upregulated by vascular endothelial growth factor (VEGF) [3][4][33,34]. Therefore, VEGF and CXCL1 factors participate together in angiogenesis. The described influence of CXCL1 on endothelial cells also implicates CXCL1 in blood vessel regeneration as CXCL1 expression in endothelial cells is increased by tissue factor, the primary initiator of blood coagulation [5][35]. CXCL1 also causes the recruitment of circulating endothelial progenitor cells [6][7][36,37], endothelial cell chemotaxis and proliferation [1][2][8][31,32,38], processes that lead to wound repair.
At the same time, CXCL1 in blood vessels can also cause disease complications. In particular, angioplasty and stenting can lead to neointima formation and to the re-stenosis of blood vessels [9][39]. This process is dependent on the production of CC motif chemokine ligand 2 (CCL2) and CXCL1 by smooth muscle cells. These chemokines induce the recruitment of Sca-1+ vascular stem/progenitor cells to blood vessel sites that have undergone angioplasty and stenting, in processes dependent on CXCR2 and CC motif chemokine receptor 2 (CCR2). The recruited Sca-1+ vascular stem/progenitor cells participate in neointima formation [9][39].
CXCL1 is also important in arteriogenesis, as its expression in vein and aortic endothelial cells is increased during increased blood pressure and thus shear stress [10][11][40,41]. This is followed by CXCR2-dependent adhesion of monocytes to endothelial cells [11][41]. These monocytes differentiate into M2 macrophages [12][42] which then secrete growth factors that act on smooth muscle cells and endothelial cells, thus enabling collateral remodeling and arteriogenesis [13][43].
2. Atherosclerosis
Atherosclerosis is an arterial disease that is characterized by inflammatory reactions in the blood vessel walls, intimal lipoprotein deposition and the formation of atherosclerotic plaques in advanced stages of the disease [14][44]. Atherosclerosis leads to atherosclerotic cardiovascular disease (CVD) which often ends in death due to myocardial infarction or ischemic stroke (it is estimated that 31% of all deaths worldwide are related to CVD) [15][45]. In this disease, the development of atherosclerosis involves macrophages. These cells, under the influence of oxidized low-density lipoprotein (oxLDL), are converted to foam cells.
CXCL1 is another element in the pathophysiology of atherosclerosis. Expression of this chemokine and CXCR2 receptor occurs in humans in the intima of atherosclerotic lesions [16][46]. oxLDL causes an increase in CXCR2 expression on peripheral blood monocytes [17][47]. Activated mast cells [18][48] and macrophages, in particular those interacting with vascular smooth muscle cells [19][49], produce CXCL1 in intima and perivascular tissue. Another source of CXCL1 in arterial vessel walls is endothelial cells where oxLDL increases the expression of CXCL1 [20][50]. At the same time, lysophosphatidic acid (LPA) may also be responsible for the production of CXCR2 ligands in arterial vessel walls [21][51]. LPA induces an increase in the release of endothelial CXCL1, or at least CXCL1/KC, the murine paralog for human CXCL1, as shown by experiments on mice [19][49]. Pro-inflammatory cytokines such as IL-1β and TNF-α are also important factors that increase CXCL1 expression in the inflamed endothelium and atherosclerotic lesions [22][52].
CXCL1 causes the recruitment of neutrophils into the atherosclerotic plaque [18][48]. These cells play an important role in atherosclerotic lesions as the recruited neutrophils secrete pro-inflammatory cytokines that participate in inflammatory responses [23][53]. Neutrophils also secrete proteases which cause plaque destabilization. CXCL1 also affects monocytes. The CXCL1→CXCR2 axis increases adhesion and arrest of monocytes to the inflamed endothelium, which leads to the recruitment of these cells to the vessel wall [16][20][21][24][25][46,50,51,54,55]. The action of CXCL1 in recruiting monocytes to early atherosclerotic lesions seems to be more important than that of CCL2 [24][25][54,55]. However, there are also studies contradicting this and showing that the aforementioned axis is only important in advanced atheromatous plaques [26][56]. CXCL1 also affects monocytes after recruitment. It increases the expression of class A scavenger receptor (SR-A) and cluster of differentiation 36 (CD36) in macrophages [22][52], both being receptors for oxLDL [27][28][57,58]. As CXCL1 increases the accumulation of oxLDL in macrophages, it increases the rate of conversion of these cells to foam cells [22][52].
CXCL1 also causes the release of matrix metalloproteinases (MMPs) from vascular smooth muscle cells, which leads to plaque destabilization (Figure 1) [22][52].
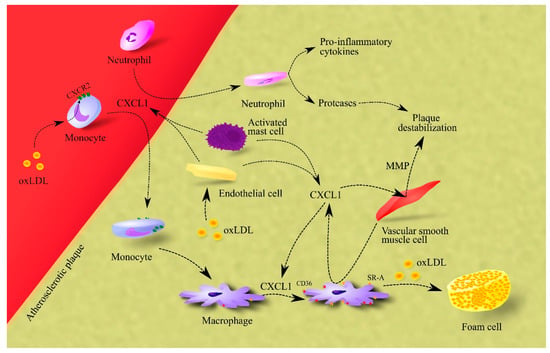
Figure 1. Importance of CXCL1 in atherosclerosis. In atherosclerotic plaque, endothelial cells, macrophages and activated mast cells are responsible for CXCL1 production. This chemokine causes the recruitment of neutrophils and monocytes to atherosclerotic plaque. The effect on monocytes is enhanced by oxLDL, which increases CXCR2 expression on these cells. After recruitment, neutrophils secrete pro-inflammatory cytokines that increase inflammation in the atherosclerotic plaque. These cells also secrete proteases that cause plaque destabilization. In turn, monocytes differentiate into macrophages which, under CXCL1, increase the expression of CD36 and SR-A, receptors for oxLDL. Therefore, CXCL1 increases the uptake of oxLDL by macrophages. This elevates the rate of foam cell formation in the atherosclerotic plaque. Additionally, CXCL1 causes an increase in the production and secretion of MMPs in vascular smooth muscle cells; these are proteases that cause plaque destabilization. Abbreviations: CXCL1–CXC motif chemokine ligand 1; CXCR2–CXC motif chemokine receptor 2; MMP–matrix metalloproteinases; oxLDL–oxidized low-density lipoprotein; SR-A–class A scavenger receptor.
Finally, CXCL1 may also participate in atherosclerosis regression [29][59]. It is responsible for the recruitment of bone-marrow endothelial progenitor cells to atherosclerotic plaque, which leads to plaque resolution.
3. Atrial Fibrillation
One of the consequences of hypertension is atrial fibrillation [30][60]. Patients with atrial fibrillation have higher levels of CXCL1 and CXCR2+ monocytes in their blood [31][61]. At the same time, atrial fibrillation patients have lower CXCL1 expression in the atrium heart tissue and higher CXCR2 expression compared to healthy subjects [32][62]. This suggests that if CXCR2 has a role in the pathogenesis of atrial fibrillation, then it is activated by a ligand other than CXCL1. Animal studies have shown that atrial fibrillation is associated with increased expression of CXCR2 ligands in the atrium and subsequent infiltration by CXCR2+ immune cells [31][61]. In particular, infiltration of monocytes in the atrium tissue leads to an increase in the number of macrophages in the atrium [31][61]. The level of CXCL1 expression in the atrium in patients with atrial fibrillation is strongly positively correlated with activated mast cells, weakly positively correlated with M2 macrophages and negatively correlated with M1 macrophages [33][63]. Mast cells may contribute to atrial fibrillation by secreting platelet-derived growth factor A (PDGF-A) [34][64]. This growth factor increases collagen production by cardiac fibroblasts which leads to atrial fibrosis and atrial fibrillation. At the same time, patients with atrial fibrillation do not experience increased infiltration of atrial tissue by mast cells [35][65], which indicates that this mechanism may not be significant in atrial fibrillation. In humans, an important role in atrial fibrillation may be played by CXCL7/pro-platelet basic protein (PPBP), a ligand of CXCR2. Expression of this chemokine in the atrium in patients with atrial fibrillation is positively correlated with infiltration of atrial tissue by neutrophils and monocytes [33][63]. In addition, CXCL7/PPBP and CXCL1 are the hub gene in atrial fibrillation with the highest number of associations [33][63].
4. Chronic Oschemic Heart Disease
Patients with chronic ischemic heart disease have elevated levels of CXCL1 expression in the blood [36][66] and heart [37][67]. In patients with ischemic heart failure, an increase in cardiac CXCL1 expression may be mediated by T helper type 17 (Th17) cells [37][67]. CXCL1 acts as a chemoattractant for bone marrow-derived endothelial precursors [38][68] which participate in vasculogenesis and thus counteract disease progression. Nevertheless, CXCR2-dependent recruitment of immune cells to the heart occurs after infarction [39][69]. The recruited immune cells cause inflammatory reactions that result in further damage to the heart.
5. Heart Failure
Elevated blood levels of CXCL1 have been observed in patients with heart failure [40][41][70,71]. This increase is correlated with the TT genotype of rs33980500 of TRAF3 Interacting Protein 2 (TRAF3IP2) [40][70]. In contrast, no increase in CXCR2 expression has been observed in the hearts of patients with heart failure [42][72]. This shows that elevated levels of CXCL1 in the blood can lead to heart failure as shown by experiments on laboratory animals. In angiotensin II-treated mice, there is an increase in CXCL1/KC expression in the heart [41][71]. It is a mouse CXCR2 ligand often equated with human CXCL1. The increased expression of CXCR2 ligands in the heart leads to the infiltration of the heart by CXCR2+ immune cells, including macrophages, neutrophils, and T cells [31][41][61,71]. The same results were obtained by studying spontaneously hypertensive rats [43][44][73,74]. In the heart, these immune cells cause cardiac hypertrophy, fibrosis, atrial fibrillation and inflammation [31][39][41][61,69,71]. Mast cellsproduce PDGF-A that causes atrial fibrosis [34][64]. In addition, macrophages cause atrial fibrosis [45][75], inflammation in heart tissue and cardiac hypertrophy [46][76]. These incremental changes in the heart inevitably lead to heart failure [47][48][77,78].
6. Hypertension
CXCL1 can also cause hypertension. People with hypertension have higher levels of CXCL1 in their blood than healthy individuals [49][79]. The mechanism of its involvement in the pathogenesis of hypertension has been studied in laboratory animals. In mice treated with angiotensin II, the resulting hypertension is dependent on the CXCL1/KC→CXCR2 axis [50][80]. In this process, increased CXCL1/KC expression in the aorta leads to infiltration of the aorta by cells with a high CXCR2 expression, in particular by macrophages [50][51][80,81]. At the same time, the infiltration by monocytes, from which macrophages are formed, may depend on activated B cells [52][82]. Macrophages have expression of angiotensin type 1 receptor (AT1R) and angiotensin type 2 receptor (AT2R) and for this reason can respond to angiotensin [53][54][83,84]. Among other things, angiotensin II causes an increase in toll-like receptor 4 (TLR4) expression which leads to an increase in pro-inflammatory cytokine production in macrophages, which results in inflammation in blood vessels [54][84]. Additionally, macrophages in the blood vessel wall under the influence of B cells produce transforming growth factor β (TGF-β), which leads to extracellular matrix remodeling, vascular stiffness and consequent hypertension [54][84].
Hypertension can cause hypertensive retinopathy. Patients with hypertensive retinopathy have higher levels of CXCL1 in their blood than patients with hypertension but without hypertensive retinopathy and healthy individuals. Studies on mice have shown that angiotensin II-induced hypertension increases CXCL1/KC expression in the retina [49][79], which increases infiltration of the retina by CXCR2+ immune cells, particularly by neutrophils and macrophages. In the retina, these cells produce ROS which leads to oxidative stress. These cells are involved in inflammation, which, together with ROS production, leads to retinopathy [49][79].
7. Sepsis
Sepsis is an infection with acute organ dysfunction that often results in death [55][85]. It is difficult to attribute to a particular organ because sepsis causes symptoms in multiple organs. In the US alone, there are 300 cases of sepsis per 100,000 people per year. Researchers have included sepsis as a cardiovascular disease due to the importance of circulation in the early stages of the sepsis course. Sepsis is closely associated with the inflammatory response which is associated with elevated blood levels of pro-inflammatory cytokines such as TNF-α, IL-1β and interleukin-6 (IL-6) [56][86]. CXCL1 levels also tend to increase in sepsis patients, with an increase close to statistical significance relative to healthy individuals (p = 0.07) [57][87]. However, in humans with sepsis, the most important CXCR2 ligand is CXCL8/IL-8, whose blood levels are significantly elevated [57][87]. In sepsis, the CXCR2 ligands have two actions. First, they cause mobilization of neutrophils from the bone marrow and infiltration of various organs by these cells. Then, neutrophils contribute to the eradication of bacterial pathogens [58][88] but also contribute to tissue damage resulting in organ dysfunction.
One of the symptoms of the described disease state is sepsis-associated encephalopathy—damage to the nervous tissue. In the brains of individuals who died from sepsis, expression of CXCL1 was significant in two out of three reported cases [59][89], although increased expression of CXCL8/IL-8 was observed in all three cases. Increased production of CXCR2 ligands is also confirmed in animal models [60][61][90,91]. CXCL1 production in the brain in sepsis is carried out by astrocytes [60][90] and microglial cells [62][92], in a process dependent on pro-inflammatory cytokines. Sepsis-associated encephalopathy is caused by the infiltration of the brain by neutrophils, in a process dependent on the expression of CXCR2 on cerebral endothelial cells [63][93]. Activation of this receptor results in the increased expression of P-selectin, vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1), which are involved in the rolling and adhesion of leukocytes and the subsequent recruitment of neutrophils to the brain [60][90]. However, the primary driver of endothelial activation may not be the CXCL1→CXCR2 axis but lipopolysaccharide (LPS) and TNF-α [63][93].
Sepsis also causes acute kidney injury (AKI). Studies on mice have shown that CXCL1/KC and CXCL2/macrophage inflammatory protein-2 (MIP-2) expression is increased in the kidney during sepsis [64][65][66][94,95,96]. CXCL1/KC is a hub gene in septic AKI in mice [66][96]. The IL-17–dependent increase in murine CXCL1/KC and CXCL2/MIP-2 expression in the kidney leads to the infiltration of the kidney by neutrophils [65][95] which contribute to kidney damage by secreting proteases, particularly neutrophil elastase [67][97]. This process occurs in patients with sepsis, as patients with renal dysfunction show an increased neutrophil-to-lymphocyte ratio [68][98]. Nevertheless, further studies are required to demonstrate which CXCR2 ligands are involved in this process in humans.
Murine sepsis is associated with the purinergic receptor P2X7 (P2X7)-dependent generation of reactive species and lipid peroxidation in the liver, leading to the apoptosis of liver cells [69][99]. Additionally in the liver, in hepatocytes, there is a P2X7-dependent increase in the expression of CXCL1/KC and CXCL2/MIP-2 as shown by experiments in mice [69][70][71][99,100,101]. This leads to infiltration of the liver by neutrophils which then cause liver injury in a process dependent on apoptotic cells in the liver [71][101]. CXCL1/KC produced by the liver also causes myeloid-derived suppressor cells (MDSC) mobilization which contributes to the control of systemic inflammation—a mechanism that may mitigate the course of sepsis [72][102]. Nevertheless, the involvement of CXCL1 in liver dysfunction requires further investigation.
Sepsis often leads to ileus, associated with TLR4 activation on myocytes in the ileum, a process which leads to an increase in CXCL1/KC expression in mice [73][103]. This chemokine contributes to reduced contraction amplitude in the ileum and consequently to the ileus [74][104]. Further studies on humans are required to demonstrate which CXCR2 ligand plays an important role in this process.