Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Vincenzo Sorrentino.
Nicotinamide Adenine Dinucleotide (NAD+) and its reduced form NADH, by regulating redox reactions and allowing the production of adenosine triphosphate (ATP), are crucial for energy metabolism and fatty acid β-oxidation (FAO) in proximal tubular epithelial cells (PTECs). In addition, by being the substrate of non-redox NAD+-consuming enzymes such as sirtuins and poly(ADP-ribose) polymerases (PARPs), NAD+ is also involved in several key molecular mechanisms for cellular homeostasis. NAD+ is present in the kidney at concentrations ranging from 0.3 to 1 mmol/kg of tissue, which is comparable to concentrations found in liver and muscle.
- NAD+
- NAD+ precursors
- premature renal aging
- chronic kidney disease
- kidney
1. NAD+ Homeostasis and Precursors
Mammals can produce NAD+ from different dietary precursors, which include the essential amino acid L-tryptophan (L-Trp), different forms of vitamin B3, and the NAD+ breakdown products that contain a pyridine ring. To date, there are at least five different pathways for NAD+ production that are grouped into (i) the de novo biosynthesis pathway from L-Trp; (ii) the Preiss-Handler pathway fueled by nicotinic acid (NA); and (iii) the amidated salvage pathways which include the salvage of nicotinamide (NAM) to nicotinamide mononucleotide (NMN), the conversion of nicotinamide riboside (NR) to NMN as well, and the conversion of the reduced form of NR, dihydronicotinamide riboside (NRH), to dihydronicotinamide mononucleotide (NMNH) (Figure 1). The kidney expresses all of the NAD+-synthesizing enzymes, most of them abundantly [24][1], and is therefore able to take advantage of all five pathways. Quantitative analysis of in vivo NAD+ fluxes with labeled precursors confirmed that the kidney is the only other organ, after the liver, that can produce and excrete NAD+, primarily as NAM [28][2]. However, the kidney generates about 5% of the total circulating NAM, so renal NAD+ production is unlikely to be a critical driver of systemic NAD+ needs. In addition, kidney and liver, as well as the other tissues, take up circulating NAM or other NAD+ precursors and “salvage” it as NAD+.
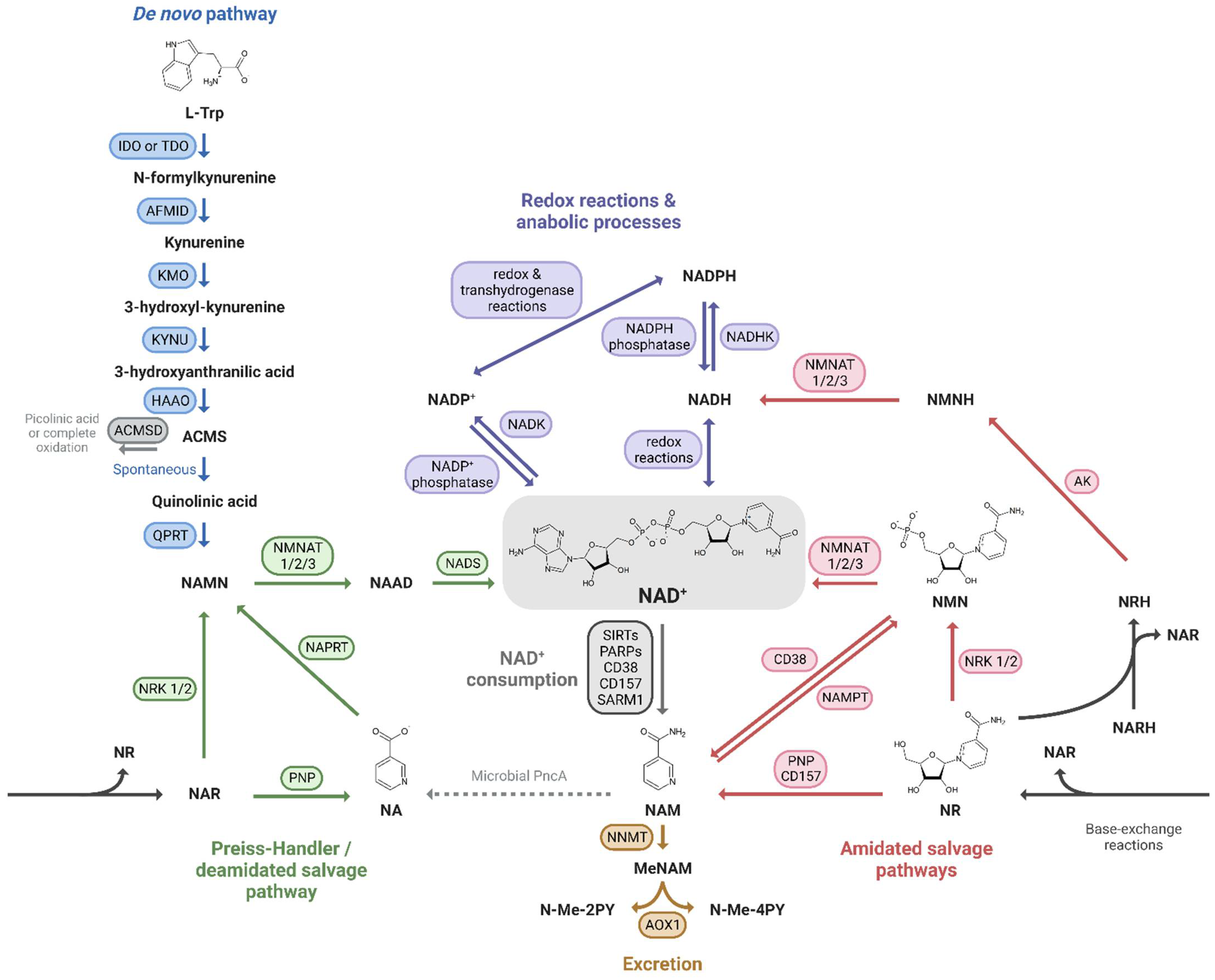
Figure 1. NAD+ precursors and biosynthesis pathways. Depiction of the three routes leading to NAD+ synthesis: (i) the de novo pathway from L-Trp (blue); (ii) the Preiss-Handler/deamidated salvage pathway from NA (green); and (iii) the amidated salvage pathways including the salvage of NAM to NMN and the conversion of NR and NRH to NMN and NMNH, respectively (red). Intermediate forms of nucleotides in each pathway and reactions linking the amidated and deamidated routes (base-exchange reactions and microbial deamidation) are indicated. The fate of NAD+ is also shown: its use in redox reactions, via NADH, and anabolic processes, via the NADP+/NADPH couple (purple), its enzymatic consumption (grey), and finally its oxidation and excretion from the body (yellow).
Even though NAD+-synthesizing enzymes are present in both the tubulointerstitial and glomerular compartments of the kidney [29][3], the focus here is on the metabolism of PTECs, given their high metabolic demand. Indeed, prior to use it, PTECs take up NAD+ and its precursors at their brush borders, through the sodium-monocarboxylate transporter SLC5A8 (SMCT) or SLC22A13 for NA and through unidentified transporters for NAM [30,31][4][5]. NMN as well as NR and its reduced form NRH are imported by equilibrative nucleoside transporters (ENTs) [27,32][6][7]. L-Trp is taken up by neutral amino acid transporters [33][8] (Figure 2). The specific biosynthetic pathways of NAD+ metabolism, its excretion and its compartmentalization are described below.
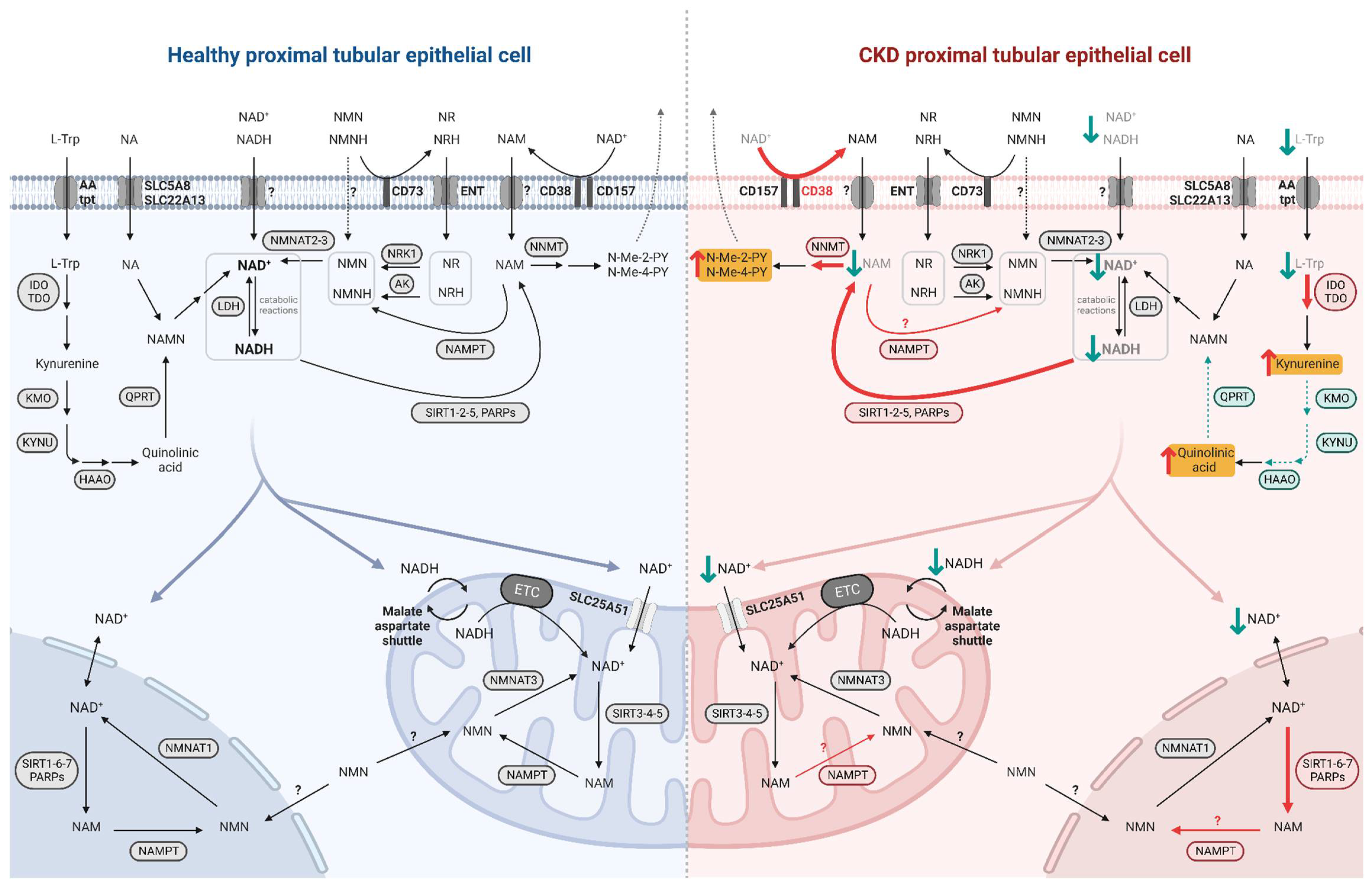
Figure 2. NAD+ metabolism in healthy and diseased PTECs. NAD+ precursors are taken up by specific transporters at the brush border of PTECs. NAD+ biosynthesis occurs primarily in the cytosol while its consumption and salvage can occur in the cytosol, nucleus, and mitochondria, involving isoforms of enzymes specific to these compartments. NAD+ moves freely between the nucleus and cytosol while it requires specific transport mechanisms to enter the mitochondria. In diseased PTEC (right side), NAD+ biosynthesis is impaired and its consumption is increased, resulting in decreased NAD+ levels and increased uremic toxin levels. Enhanced reactions and accumulated metabolites in CKD and premature renal aging are indicated by red arrows, with uremic toxins highlighted in yellow; impaired reactions and decreased metabolites are indicated by green arrows.
1.1. De Novo Pathway
The nine-step de novo pathway allows the synthesis of kynurenine from L-Trp, which can be processed into various signaling molecules or give rise to amino-β-carboxymuconate-ε-semialdehyde (ACMS). ACMS can in turn be processed by ACMS decarboxylase (ACMSD) or undergo spontaneous cyclization to form quinolinic acid [34][9]. QA can then only be converted by the enzyme quinolate phosphoribosyl transferase (QPRT) to nicotinic acid mononucleotide (NAMN), which then feeds into the Preiss-Handler pathway.
The kidney is, along with the liver, one of two organs capable of converting L-Trp to NAD+, and both tissues appear to prefer this precursor to NA based on quantitative analysis of in vivo NAD+ flux by Liu et al. [28][2]. Loss-of-function mutations in two de novo enzymes, 3-hydroxyanthranilate 3,4-dixoygenase (HAAO) and kynureninase (KYNU), are associated with multiple congenital malformations, including major renal abnormalities, resulting in a reduction in circulating NAD+ levels in patients [35][10].
1.2. Preiss-Handler Pathway
The Preiss-Handler pathway, or deamidated salvage pathway, consists of three steps [36][11]: nicotinic acid phosphoribosyltransferase (NAPRT) produces NAMN from the dietary NA or utilizes NAMN from the L-Trp processing, and nicotinamide mononucleotide adenylyltransferases (NMNATs) catalyze the addition of the adenylyl moiety from ATP to form nicotinic acid adenine dinucleotide (NAAD), which is finally amidated to NAD+ by the NAD+ synthetase (NADS) [37][12].
In mice, NA has been shown to increase renal NAD+ levels, but less efficiently than NAM [38][13]. Indeed, the deamidated route accounts for a minority of the NAD+ synthesized in mice kidneys, as evidenced by the low activity of NADS compared to NMNATs, which are the last-step enzymes in both routes [39][14]. While NA and its derivatives have already been used clinically for CKD, primarily because of its phosphate-lowering properties (see Section 3.2) the actual physiological importance of the Preiss-Handler pathway in human kidney NAD+ homeostasis remains to be assessed.
1.3. Amidated Salvage Pathways
The amidated salvage pathways include recycling of NAM (sometimes referred to exclusively as the salvage pathway [40][15]), that is one of the breakdown products of the NAD+-consuming enzymes, and the phosphorylation of dietary NAD+ precursor NR and its reduced form NRH. The enzymes that catalyze these three reactions are NAMPT [41][16], NR kinases (NRKs) [42][17] and adenosine kinase (AK) [27][6], respectively, and the product is NMN/NMNH, which is then converted to NAD+/NADH by NMNATs.
NAMPT is the rate-limiting enzyme for the recycling of NAM to NAD+ [43][18] and is controlled by the circadian clock machinery, as are the intracellular NAD+ levels [44,45][19][20]. Homozygous Nampt KO mice are embryonically lethal, and heterozygous animals have lower NAD+ levels in tissues that do not generate de novo NAD+ [46][21], but also in the liver, a tissue with an active de novo pathway [47][22], suggesting that NAM salvaging is the major pathway for NAD+ production in mammals.
NRK1 is essential for the maintenance of energy homeostasis and catalyzes the rate-limiting step for the efficient utilization of exogenous NR and NMN [48][23]. Indeed, it seems that NMN requires to be dephosphorylated by CD73 before entering the cells as NR where it can be phosphorylated back to NMN [49][24], even though a specific NMN transporter highly expressed in the small intestine has been recently discovered [50][25]. In addition, NRKs can phosphorylate nicotinic acid riboside (NAR), the deamidated form of NR, to generate NAMN that can thus enter the Preiss-Handler pathway [51][26]. Both NR and NAR can be hydrolyzed by purine nucleoside phosphorylase (PNP) to NAM and NA, respectively [52][27].
Recently, the reduced form of NR, NRH, has gained interest as a very potent precursor of NAD+, especially in the kidney [27,53][6][28]. NMNH has also been shown to increase NAD+ levels after a dephosphorylation step to NRH prior to its incorporation into the intracellular space [54][29]. A final reduced precursor, dihydronicotinic acid riboside (NARH) can act synergistically with nicotinamide riboside (NR) to increase NAD+ through their conversion to NRH [26][30]. The greater effectiveness of NRH compared to NR as an NAD+ booster could be based on both the higher enzymatic phosphorylation rate of AK compared to NRK1, and the fact that NRH is not or less degraded to NAM in plasma, which is the case for NR [27][6].
1.4. Phosphorylation to NADP+
Two other fates of the NAD+ molecule, if it is not consumed and recycled or reduced, are to be noted: its phosphorylation and its excretion. The first, which represents about 10% of total NAD(H) [28][2], involves NAD+ kinase (NADK) to produce NADP+ [55][31]. The transhydrogenase activity of different enzymes leads to a rapid conversion to NADPH, both in the cytosol and in the mitochondria, favoring the reduced form of the NADP+/NADPH couple as opposed to NAD+/NADH [56,57][32][33]. In turn, NADP+ can be converted back to NAD+ by NADP+ phosphatases [58][34].
1.5. Excretion
The last metabolic route of NAD+ homeostasis is the excretion of its metabolites from the body via the urine. The enzyme nicotinamide N-methyltransferase (NNMT) catalyzes the methylation of NAM to form methylated NAM (MeNAM) [59][35] that can be further oxidized to N-methyl-2-pyridone-5-carboxamide (N-Me-2PY) and N-methyl-4-pyridone-3-carboxamide (N-Me-4PY) [60][36]. In mammals, NNMT expression and activity appear to increase during aging and age-associated diseases without a clear explanation of its relevance in the healthy state or during aging pathogenesis [22,61,62,63][37][38][39][40]. In contrast, interestingly, the NNMT homolog in the short-lived nematode Caenorhabditis elegans, ANMT-1, extends lifespan. This is due to reactive oxygen species (ROS) generated by an aldehyde oxidase using MeNAM as a substrate, resulting in a mitohormesis signal [64][41].
1.6. Compartmentalization
NAD+ can move freely between the intracellular compartments of cytosol, nucleus, and mitochondria (Figure 2). The recent discovery of a mammalian mitochondrial NAD+ transporter [65][42] has overturned the long-held theory that mitochondria rely only on imported NMN, salvaged by the specific enzyme NMNAT3 [66,67][43][44]. However, mitochondrial NAD+ levels and half-life differ from the nucleo-cytosolic pool and appear to be independently regulated [68][45]. While in most tissues and cells NAD+ appears to be more present and stable in mitochondria [69][46], in hepatocytes, the majority of NAD+ is cytosolic [70][47] which is also consistent with the subcellular localization of the de novo pathway enzymes in liver [24][1]. Given that the de novo pathway has also been described in the kidney, the renal NAD+ pool may be present in both mitochondria and cytosol, yet to be determined. Finally, the subcellular distribution of NAD+ seems to vary according to the metabolic needs of the cells by locally activating specific NAD+-consuming enzymes and not to disrupt the activity of others [71][48]. Detailed description of the subcellular distribution of NAD+ and its biosynthesis enzymes has been reviewed by Katsyuba et al. [24][1].
2. NAD+ Cellular Roles
2.1. Redox Co-Factor
NAD+ is an essential cellular metabolite for every cell. First described in 1906 as a crucial component for yeast fermentation [72][49], it took 30 years to understand the ability of this thermostable cofactor to accept a hybrid ion and transfer it to molecules [73][50].
In both the cytosol and mitochondria, NAD+ acts as an electron acceptor in several catabolic reactions, including glycolysis, the tricarboxylic acid (TCA) cycle, FAO and alcohol metabolism [74][51]. NAD+ is regenerated from its reduced form NADH by lactate dehydrogenase (LDH), which catalyzes the reduction of pyruvate to lactate, primarily under anaerobic conditions, or by mitochondrial oxidative phosphorylation (OXPHOS) complex I which provides electrons through the electron transport chain (ETC) to ultimately synthesize ATP in the presence of oxygen. Under aerobic conditions, to allow OXPHOS reactions to occur at the mitochondrial inner membrane, the NADH molecules produced in the cytosol transfer into the mitochondria with the malate/aspartate shuttle.
The NADP+/NADPH pair, on the other hand, is mainly required for anabolic reactions, such as the pentose phosphate pathway and the synthesis of fatty acids and steroids [75][52]. In addition, NADPH plays a role in oxidative mechanisms in renal diseases, both by being used by NADPH oxidase for the respiratory burst, i.e., the rapid production of ROS for immunological defense, but also for the reduction of glutathione to detoxify oxidative stress [76][53].
2.2. Enzymatic Substrate
In the 2000s, NAD+ regained much attention by being described as a substrate for metabolic and aging coordinating enzymes, the sirtuins [77][54]. Seven mammalian sirtuins have been identified, orthologs of the yeast silent information regulator Sir2 which promotes longevity through transcriptional hyper-silencing [78][55]. Sirtuins have been proposed as the key mediators of caloric restriction-induced life extension [79,80][56][57]. Sirtuins localize to distinct subcellular compartments (both nucleus and cytosol for SIRT1, nucleus for SIRT6, nucleolus for SIRT7, mitochondria for SIRT3, SIRT4, and SIRT5, and cytosol for SIRT2 and SIRT5) and differ in their enzymatic activities and targets. Sirtuins can deacetylate lysine residues of various proteins, including histones H1, H3, and H4, to condense chromatin and induce gene silencing, as well as transcription factors such as peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha, FoxO1, nuclear factor-kappa B (NF-κB), and p53 to modulate their activity and thus regulate metabolism. In addition, some sirtuins can deacylate other post-translational modifications (succinylation, malonylation, and fatty acid acylation). These reactions result in the production of the deacetylated substrate, NAM and 2-O-acyl-ADP-ribose as end products. Finally, SIRT4 and SIRT6 have mono-ADP-ribosyl transferase activity, also releasing NAM [81,82][58][59]. Interestingly, NAM non-competitively binds and thus feedback-inhibits sirtuins at least in the simple model Saccharomyces cerevisiae, underscoring the importance of NAD+ metabolites fluxes in cells [83,84][60][61].
In the kidney tissue, the most studied sirtuins are SIRT1 and SIRT3, with a recent interest in SIRT6. SIRT1 and SIRT3 are the only two sirtuins whose Km value can exceed the physiological range of NAD+ in the body (~300–700 μM), meaning that their activity is highly dependent on NAD+ availability [24][1]. SIRT1 is a key factor in mitochondrial quality maintenance, by promoting mitochondrial biogenesis through PGC1α deacetylation and the removal of defective mitochondria by mitophagy [85][62]. In the kidney, SIRT1 is involved in important signaling pathways of metabolic homeostasis and stress responses such as mitochondrial biogenesis, but also apoptosis of PTECs, autophagy and inflammation, and ultimately plays a role in renal fibrosis [86][63]. Homozygous Sirt1 knockout (KO) mice exhibit severe developmental defects [87][64] whereas heterozygous Sirt1 KO mice show more fibrosis in the model of unilateral ureteral obstruction (UUO)-induced CKD [88][65]. For diabetic nephropathy, PT-specific Sirt1 transgenic mice showed prevention of albuminuria and glomerular changes in streptozotocin (STZ)-treated and db/db mice while PT-specific Sirt1 KO mice showed worsening of the same parameters in STZ-treated mice [89][66]. The mitochondrial SIRT3 regulates lipid metabolism and the antioxidant system [90[67][68],91], and Sirt3 KO mice exhibit hyper-acetylated mitochondrial proteins in PTECs and severe renal fibrosis [92][69]. Finally, SIRT6 plays an important role against renal fibrosis. Its upregulation after UUO or ischemia/reperfusion injury (IRI) has been mechanistically explained in PTECs by its role in modulating the Wnt/β-catenin signaling pathway [93][70]. PT-specific Sirt6 KO mice show tubular basement membrane (TBM) thickening and collagen deposition, associated with the upregulation of the profibrogenic gene TIMP-1 [94][71]. Importantly, the phenotype is similar to that of Nampt KO mice, suggesting a convergent mechanism between loss of SIRT6 and dysfunction of NAD+ biosynthesis [94][71]. Several natural and synthetic sirtuin-activating compounds (STACs) have shown beneficial effects in preclinical models of kidney disease [95][72]. Resveratrol, the most widely used natural STAC, enhanced SIRT1 activity and ameliorated tubular fibrosis, in the 5/6 nephrectomy [96][73] and the UUO models [97][74]. The mechanism in PTECs was found to be via deacetylation of Smad 3 [97][74] or Smad 4 [98][75], important transcription factors of the transforming growth factor-beta (TGFβ) pathway. Similarly, honokiol, a STAC activating SIRT3, protected against UUO-induced tubular fibrosis through the regulation of the NF-κB/TGF-β1/Smad signaling pathway and mitochondrial dynamics [99][76].
NAD+ is also a substrate for poly(ADP-ribose) polymerases (PARPs) that transfer its ADPr moiety to nuclear proteins, thereby releasing NAM. The target proteins end up with long chains of ADPr homopolymers; this post-translational modification is called PARylation and is reversible. The PARP family, which has 17 members in humans and 16 in mice, plays a key role in the maintenance of genomic stability [100][77]. Specifically, PARylation of histones and other proteins, including PARP itself, enables the recruitment and activation of DNA repair enzymes at the damaged site [101][78]. PARP 1 and PARP 2 account for about 90% and 10% of the NAD+ used by the PARP family, respectively [102][79], which is about one-third of the total NAD+ turnover under basal conditions but can be up to 60% under overt DNA damage [28][2], depleting intracellular NAD+ levels. The low Km of PARPs for NAD+ suggests that their enzymatic activity may not be limited when NAD+ levels decrease [24][1], whereas it may become limiting for sirtuins, as observed after genotoxic stress or during aging and which partly explains the altered mitochondrial metabolism resulting from these conditions [103][80]. PARP 1/2 inhibition could therefore alleviate diseases with PARP overactivation by increasing the availability of NAD+ for sirtuins [104][81]. Consistently, Parp1 KO mice are more resistant to functional and histological damages following ischemic AKI [105][82], UUO [106][83], and STZ-induced diabetic nephropathy [107][84] than their wildtype littermates. The mechanism converges towards a decrease neutrophil infiltration and reduction in expression of proinflammatory proteins [105,106][82][83].
Beyond sirtuins and PARPs, NAD+ is also consumed by CD38, CD157 and SARM1. The cell surface protein CD38 and its homologue CD157 are multifunctional enzymes and cell receptors important for the immune system. First identified on T cells and on bone marrow stromal cells, CD38 and CD157, respectively, are now known to be expressed in several tissues, including the kidney [108,109][85][86]. Both are primarily NAD+ glycohydrolases (with a low Km [24][1]), have an inefficient ADP-ribosyl cyclase activity, and can also mediate base-exchange reactions to produce nicotinic acid adenine dinucleotide (phosphate) (NAAD(P)) and NAM from NAD(P) and NA, or NAR and NAM from NR and NA, respectively for CD38 and CD157 [108,110][85][87]. The three end products APDr, cyclic ADPr (cADPr) and NAAD(P) are second messengers that mobilize intracellular calcium [111][88]. CD38 and CD157 are mainly ectoenzymes catabolizing NAD+ in the extracellular environment, but CD38 may also have its catalytic site facing the intracellular environment [112][89]. Moreover, CD38 has also been identified as an ecto-NMNase [113,114][90][91] and CD157 as an ecto-NRase [108[85][92],115], degrading NAD+ precursors before their entry into cells. The enzymatic activities of CD38 and CD157 help explain the decrease in NAD+ levels associated with their upregulated expression in aging tissues [61,113,114][38][90][91]. Pharmacological inhibition of CD38 in old mice increased NAD+ levels in several tissues, including skeletal muscle, liver, and spleen, but they were not measured in the kidney [116][93]. Finally, SARM1, primarily expressed in neurons, also possesses NAD(P)+ glycohydrolase, ADP-ribosyl cyclase, and base exchange activities [117,118][94][95]. NAD+ degradation by SARM1 has been linked to axonal degeneration [119][96]. Although SARM1 appears to be expressed in the kidney, its functionality has not yet been explored [120][97].
3. NAD+ Alterations in the Diseased Kidney
The first link between NAD+ levels and health was established in 1937, when Pellagra disease was found to be caused by dietary niacin deficiency [121][98]. Since then, there have been numerous reports describing a decline in NAD+ levels in many pathological conditions but especially in the context of aging [122][99]. This decline has been associated with several established molecular hallmarks of aging including mitochondrial dysfunction, DNA damage, cellular senescence, and loss of proteostasis, and may be causally linked to several pathologies, including premature renal aging and kidney diseases [122][99]. Indeed, the kidneys of old mice have lower levels of NAD+, correlating with increased expression of fibrotic genes and impaired mitochondrial respiration [123][100], as well as decreased expression of SIRT1 linked to greater susceptibility to kidney injury [23][101]. In humans, the decrease of NAM and NMN circulating metabolites correlates with CKD staging [22][37]. Systemic loss of NAD+ levels is caused by (i) an imbalance in synthesis, due to altered expression of NAD+-homeostasis enzymes, and (ii) increased NAD+ consumption [124][102] (Figure 2).
3.1. Impaired NAD+ Production
Since the study by Poyan-Mehr et al. revealed that the de novo NAD+ biosynthetic pathway is impaired in ischemic AKI, due to reduced expression and activity of QPRT [125][103] (Figure 1), the defective de novo pathway has been linked to AKI-to-CKD progression, a premature renal aging phenotype and staging of CKD in humans. The persistence of a high urinary quinolinate/tryptophan ratio (uQ/T), an indirect indicator of impaired QPRT activity that is an early marker of AKI, has been shown to predict progression to CKD in the clinic [126][104]. In addition, the expression of the enzymes kynurenine 3-monooxygenase (KMO), KYNU, HAAO, and QPRT is downregulated in a stage-dependent manner in human tubulointerstitial and glomerular compartments [29][3]. A genome-wide association study (GWAS) identified variations in the KMO gene associated with albuminuria [127][105], and a follow-up study confirmed that KMO expression is downregulated in human and mouse diabetic kidney glomeruli. Furthermore, knockdown of Kmo expression in zebrafish by morpholinos injection into fertilized eggs and KO of Kmo in mice each led to a spontaneous proteinuria phenotype and podocyte foot process effacement [128][106]. These results correlate with a decrease in de novo enzymes expression in preclinical models of CKD; mRNA expression of Afmid, Kmo, Kynu, Haao and Qprt, is downregulated in UUO- and POD-ATTAC-injured mice (model of progressive secondary tubulointerstitial lesions induced by high-range glomerular proteinuria) [29][3]. Protein expression of QPRT is also decreased in models of 5/6 nephrectomy and adenine-induced nephropathy [129][107]. Concurrently, in patients with CKD, the activity of the enzymes catalyzing the first step of the de novo pathway, the conversion of L-Trp to N-formylkynurenine, namely tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO) is increased [29[3][108],130], as IDO is known to be sensitive to chronic inflammation, a feature of premature renal aging [131][109]. Increased TDO and IDO activity results in decreased circulating L-Trp levels and significant release of kynurenine metabolites such as kynurenine, kynurenic acid, and quinolinic acid [132,133,134[110][111][112][113],135], which are classified as protein-bound uremic toxins and linked to vascular complications [136][114]. In kidney transplant patients, for whom IRI is an inevitable consequence of transplantation, serum and urine kynurenine/tryptophan ratios are increased and correlate with graft rejection. Indeed, higher IDO immunostaining has been detected in biopsies from rejected kidneys, particularly in PTECs [137][115]. Importantly, it is also possible to improve NAD+ de novo biosynthesis, as Tran et al. showed that activation of PGC-1α can stimulate NAD+ de novo biosynthesis and metabolic flexibility in renal tubules, resulting in less fat accumulation and protection against worsening of kidney injury [138][116]. Conversely, NAD+ regulates PGC1α through its deacetylation by SIRT1.
NAMPT, the major enzyme of the amidated salvage pathway, is significantly decreased in multiple tissues during aging and metabolic disorders [139,140][117][118]. In the context of kidney disease, downregulation of NAMPT expression correlates with albuminuria and a fibrotic phenotype in STZ-treated diabetic mice [94][71] and has also been detected in the cisplatin-induced AKI rodent model [141,142][119][120]. Chronic inflammation mediated by inflammatory cytokines and oxidative stress is thought to be the reason why NAMPT expression is reduced during aging and age-related diseases [143][121]. However, surprisingly, opposing studies have revealed that glomerular cells (mesangial cells and podocytes) as well as PTECs increase NAMPT expression in diabetic nephropathy rat models, Otsuka Long-Evans Tokushima fatty (OLETF) rats [144][122] and STZ-treated rats [145,146][123][124]. In these studies, exogenous NAMPT has a proinflammatory effect on mesangial cells, explained by increased glucose uptake [147][125] and activation of NF-κB [145][123], while endogenous NAMPT protects PTECs from cell death in a proinflammatory environment [146][124]. This could be explained by the higher concentrations of exogenous/circulating NAMPT, which could also be released from fat and liver [146][124], and its insulin-mimicking effect further inducing glucotoxicity in the kidney cells of diabetic animals.
Additionally, Nmnat1 and Nrk1 are downregulated in the UUO model, whereas Nmnat1, Nmnat3 and Nrk1 are down-regulated in the POD-ATTAC model [29][3]. Boosting the salvage of NAM alleviates the STZ-induced diabetic nephropathy phenotype as the albuminuria of PT-specific Nampt-overexpressing transgenic mice is ameliorated [94][71].
Lastly, NNMT has been observed to be upregulated during CKD in humans and in the UUO mice model, resulting in decreased NAM salvage [22][37]. The final metabolites N-Me-2PY and N-Me-4PY are considered uremic toxins present at extremely high serum concentrations in CKD patients compared to healthy subjects [148][126].
3.2. Accelerated NAD+ Consumption
During aging and under pathological conditions, including but not limited to those leading to CKD, PARPs and CD38 are increased in expression and turnover activity, leading to accelerated NAD+ consumption [122,149][99][127].
In rats subjected to renal IRI, Parp1 expression was increased in damaged renal proximal tubules starting 6–12 h after reperfusion and maintained for several days [150][128]. After 10 days of UUO in mice, PARP1 expression and activation in the kidney were strongly increased by 6- and 13-fold, respectively [106][83]. The poly(ADP-ribosyl) protein content of the renal cortex was increased by 18% in STZ-treated diabetic mice compared with controls [107][84]. Clinically, PARP-1 expression in kidney transplant biopsies appears to correlate with cold ischemia time, acute tubular necrosis and renal function at biopsy [151,152][129][130].
CD38 NAD+ glycohydrolase is upregulated in both PTECs and glomeruli of Zucker diabetic fatty rat’s kidneys and is associated with a decrease in the NAD+/NADH ratio in the rodent renal cortex and in mitochondrial protein extracts isolated from the cortex [153,154][131][132]. In a lipopolysaccharide (LPS)-induced acute kidney injury model, CD38 expression was markedly increased on renal macrophages, resulting in their M1 polarization [155][133]. Currently, there is no clinical observation of increased CD38 expression or activity in the context of kidney disease yet; further studies will be needed to confirm the relevance of this NAD+ consumption pathway in the pathogenesis of human renal aging and CKD.
References
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31.
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5.
- Faivre, A.; Katsyuba, E.; Verissimo, T.; Lindenmeyer, M.; Rajaram, R.D.; Naesens, M.; Heckenmeyer, C.; Mottis, A.; Feraille, E.; Cippa, P.; et al. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 60–68.
- Kempson, S.A.; Colon-Otero, G.; Ou, S.Y.; Turner, S.T.; Dousa, T.P. Possible role of nicotinamide adenine dinucleotide as an intracellular regulator of renal transport of phosphate in the rat. J. Clin. Investig. 1981, 67, 1347–1360.
- Gopal, E.; Fei, Y.J.; Miyauchi, S.; Zhuang, L.; Prasad, P.D.; Ganapathy, V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by slc5a8, a member of the Na/glucose co-transporter gene family. Biochem. J. 2005, 388, 309–316.
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Perez, R.; Sanchez-Garcia, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol. Metab. 2019, 30, 192–202.
- Nikiforov, A.; Dolle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011, 286, 21767–21778.
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789.
- Ikeda, M.; Tsuji, H.; Nakamura, S.; Ichiyama, A.; Nishizuka, Y.; Hayaishi, O. Studies on the Biosynthesis of Nicotinamide Adenine Dinucleotide. Ii. A Role of Picolinic Carboxylase in the Biosynthesis of Nicotinamide Adenine Dinucleotide from Tryptophan in Mammals. J. Biol. Chem. 1965, 240, 1395–1401.
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552.
- Preiss, J.; Handler, P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 1958, 233, 488–492.
- Hara, N.; Yamada, K.; Terashima, M.; Osago, H.; Shimoyama, M.; Tsuchiya, M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 2003, 278, 10914–10921.
- Collins, P.B.; Chaykin, S. The management of nicotinamide and nicotinic acid in the mouse. J. Biol. Chem. 1972, 247, 778–783.
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014, 9, e113939.
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53.
- Rongvaux, A.; Shea, R.J.; Mulks, M.H.; Gigot, D.; Urbain, J.; Leo, O.; Andris, F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 2002, 32, 3225–3234.
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502.
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763.
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657.
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654.
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375.
- Dall, M.; Trammell, S.A.J.; Asping, M.; Hassing, A.S.; Agerholm, M.; Vienberg, S.G.; Gillum, M.P.; Larsen, S.; Treebak, J.T. Mitochondrial function in liver cells is resistant to perturbations in NAD(+) salvage capacity. J. Biol. Chem. 2019, 294, 13304–13326.
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103.
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949.
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57.
- Tempel, W.; Rabeh, W.M.; Bogan, K.L.; Belenky, P.; Wojcik, M.; Seidle, H.F.; Nedyalkova, L.; Yang, T.; Sauve, A.A.; Park, H.W.; et al. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol. 2007, 5, e263.
- Belenky, P.; Christensen, K.C.; Gazzaniga, F.; Pletnev, A.A.; Brenner, C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Biol. Chem. 2009, 284, 158–164.
- Yang, Y.; Mohammed, F.S.; Zhang, N.; Sauve, A.A. Dihydronicotinamide riboside is a potent NAD(+) concentration enhancer in vitro and in vivo. J. Biol. Chem. 2019, 294, 9295–9307.
- Zapata-Perez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Canto, C.; Lopez-Leonardo, C.; McIntyre, R.L.; et al. Reduced nicotinamide mononucleotide is a new and potent NAD(+) precursor in mammalian cells and mice. FASEB J. 2021, 35, e21456.
- Ciarlo, E.; Joffraud, M.; Hayat, F.; Giner, M.P.; Giroud-Gerbetant, J.; Sanchez-Garcia, J.L.; Rumpler, M.; Moco, S.; Migaud, M.E.; Canto, C. Nicotinamide Riboside and Dihydronicotinic Acid Riboside Synergistically Increase Intracellular NAD(+) by Generating Dihydronicotinamide Riboside. Nutrients 2022, 14, 2752.
- Lerner, F.; Niere, M.; Ludwig, A.; Ziegler, M. Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun. 2001, 288, 69–74.
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246.
- Hanukoglu, I.; Rapoport, R. Routes and regulation of NADPH production in steroidogenic mitochondria. Endocr. Res. 1995, 21, 231–241.
- Kawai, S.; Murata, K. Structure and function of NAD kinase and NADP phosphatase: Key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci. Biotechnol. Biochem. 2008, 72, 919–930.
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840.
- Felsted, R.L.; Chaykin, S. N1-methylnicotinamide oxidation in a number of mammals. J. Biol. Chem. 1967, 242, 1274–1279.
- Takahashi, R.; Kanda, T.; Komatsu, M.; Itoh, T.; Minakuchi, H.; Urai, H.; Kuroita, T.; Shigaki, S.; Tsukamoto, T.; Higuchi, N.; et al. The significance of NAD + metabolites and nicotinamide N-methyltransferase in chronic kidney disease. Sci. Rep. 2022, 12, 6398.
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283.
- Kim, H.C.; Mofarrahi, M.; Vassilakopoulos, T.; Maltais, F.; Sigala, I.; Debigare, R.; Bellenis, I.; Hussain, S.N. Expression and functional significance of nicotinamide N-methyl transferase in skeletal muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 797–805.
- Gokarn, R.; Solon-Biet, S.M.; Cogger, V.C.; Cooney, G.J.; Wahl, D.; McMahon, A.C.; Mitchell, J.R.; Mitchell, S.J.; Hine, C.; de Cabo, R.; et al. Long-term Dietary Macronutrients and Hepatic Gene Expression in Aging Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1618–1625.
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700.
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179.
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341.
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107.
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 2016, 352, 1474–1477.
- Di Lisa, F.; Ziegler, M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 2001, 492, 4–8.
- Tischler, M.E.; Friedrichs, D.; Coll, K.; Williamson, J.R. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 1977, 184, 222–236.
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223.
- Harden, A.; Young, W. The alcoholic ferment of yeast-juice. Proc. R. Soc. Lond. 1906, 77, 405–420.
- Warburg, O.; Christian, W. Pyridine, the hydrogen transfusing component of fermentative enzymes. Helv. Chim. Acta 1936, 19, 79–88.
- Krebs, H.A.; Veech, R.L. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv. Enzym. Regul. 1969, 7, 397–413.
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206.
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox. Signal. 2016, 25, 119–146.
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800.
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580.
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085.
- Sinclair, D.A. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 2005, 126, 987–1002.
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238.
- Jiang, H.; Khan, S.; Wang, Y.; Charron, G.; He, B.; Sebastian, C.; Du, J.; Kim, R.; Ge, E.; Mostoslavsky, R.; et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113.
- Anderson, R.M.; Bitterman, K.J.; Wood, J.G.; Medvedik, O.; Sinclair, D.A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003, 423, 181–185.
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107.
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95.
- Kong, L.; Wu, H.; Zhou, W.; Luo, M.; Tan, Y.; Miao, L.; Cai, L. Sirtuin 1: A Target for Kidney Diseases. Mol. Med. 2015, 21, 87–97.
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799.
- He, W.; Wang, Y.; Zhang, M.Z.; You, L.; Davis, L.S.; Fan, H.; Yang, H.C.; Fogo, A.B.; Zent, R.; Harris, R.C.; et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J. Clin. Investig. 2010, 120, 1056–1068.
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504.
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125.
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267.
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021, 12, 847.
- Cai, J.; Liu, Z.; Huang, X.; Shu, S.; Hu, X.; Zheng, M.; Tang, C.; Liu, Y.; Chen, G.; Sun, L.; et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking beta-catenin target gene expression. Kidney Int. 2020, 97, 106–118.
- Muraoka, H.; Hasegawa, K.; Sakamaki, Y.; Minakuchi, H.; Kawaguchi, T.; Yasuda, I.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Role of Nampt-Sirt6 Axis in Renal Proximal Tubules in Extracellular Matrix Deposition in Diabetic Nephropathy. Cell Rep. 2019, 27, 199–212.e195.
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809.
- Huang, X.Z.; Wen, D.; Zhang, M.; Xie, Q.; Ma, L.; Guan, Y.; Ren, Y.; Chen, J.; Hao, C.M. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell Biochem. 2014, 115, 996–1005.
- Li, J.; Qu, X.; Ricardo, S.D.; Bertram, J.F.; Nikolic-Paterson, D.J. Resveratrol Inhibits Renal Fibrosis in the Obstructed Kidney: Potential Role in Deacetylation of Smad3. Am. J. Pathol. 2010, 177, 1065–1071.
- Xiao, Z.; Chen, C.; Meng, T.; Zhang, W.; Zhou, Q. Resveratrol attenuates renal injury and fibrosis by inhibiting transforming growth factor-β pathway on matrix metalloproteinase 7. Exp. Biol. Med. 2016, 241, 140–146.
- Quan, Y.; Park, W.; Jin, J.; Kim, W.; Park, S.K.; Kang, K.P. Sirtuin 3 Activation by Honokiol Decreases Unilateral Ureteral Obstruction-Induced Renal Inflammation and Fibrosis via Regulation of Mitochondrial Dynamics and the Renal NF-κBTGF-β1/Smad Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 402.
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219.
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621.
- Bai, P.; Canto, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295.
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441.
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468.
- Zheng, J.; Devalaraja-Narashimha, K.; Singaravelu, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 2005, 288, F387–F398.
- Kim, J.; Padanilam, B.J. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2011, 301, F450–F459.
- Shevalye, H.; Maksimchyk, Y.; Watcho, P.; Obrosova, I.G. Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency alleviates diabetic kidney disease. Biochim. Biophys. Acta 2010, 1802, 1020–1027.
- Yaku, K.; Palikhe, S.; Izumi, H.; Yoshida, T.; Hikosaka, K.; Hayat, F.; Karim, M.; Iqbal, T.; Nitta, Y.; Sato, A.; et al. BST1 regulates nicotinamide riboside metabolism via its glycohydrolase and base-exchange activities. Nat. Commun. 2021, 12, 6767.
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217.
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187.
- Ernst, I.M.; Fliegert, R.; Guse, A.H. Adenine Dinucleotide Second Messengers and T-lymphocyte Calcium Signaling. Front. Immunol. 2013, 4, 259.
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67.
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139.
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarrago, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304.
- Preugschat, F.; Carter, L.H.; Boros, E.E.; Porter, D.J.; Stewart, E.L.; Shewchuk, L.M. A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch. Biochem. Biophys. 2014, 564, 156–163.
- Tarrago, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095.
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 2022, 25, 103812.
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e1335.
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 2015, 348, 453–457.
- Mink, M.; Fogelgren, B.; Olszewski, K.; Maroy, P.; Csiszar, K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/beta-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics 2001, 74, 234–244.
- Elvehjem, C.A.; Madden, R.J.; Strong, F.M.; Woolley, D.W. RELATION OF NICOTINIC ACID AND NICOTINIC ACID AMIDE TO CANINE BLACK TONGUE. J. Am. Chem. Soc. 1937, 59, 1767–1768.
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141.
- Cercillieux, A.; Ratajczak, J.; Joffraud, M.; Sanchez-Garcia, J.L.; Jacot, G.; Zollinger, A.; Métairon, S.; Giroud-Gerbetant, J.; Rumpler, M.; Ciarlo, E.; et al. Nicotinamide riboside kinase 1 protects against diet and age-induced pancreatic β-cell failure. Mol. Metab. 2022, 66, 101605.
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352.
- Chini, C.C.S.; Tarrago, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74.
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359.
- Bignon, Y.; Rinaldi, A.; Nadour, Z.; Poindessous, V.; Nemazanyy, I.; Lenoir, O.; Fohlen, B.; Weill-Raynal, P.; Hertig, A.; Karras, A.; et al. Cell stress response impairs de novo NAD+ biosynthesis in the kidney. JCI Insight 2022, 7, e153019.
- Zhang, W.; Korstanje, R.; Thaisz, J.; Staedtler, F.; Harttman, N.; Xu, L.; Feng, M.; Yanas, L.; Yang, H.; Valdar, W.; et al. Genome-wide association mapping of quantitative traits in outbred mice. G3 Genes Genomes Genet. 2012, 2, 167–174.
- Korstanje, R.; Deutsch, K.; Bolanos-Palmieri, P.; Hanke, N.; Schroder, P.; Staggs, L.; Brasen, J.H.; Roberts, I.S.; Sheehan, S.; Savage, H.; et al. Loss of Kynurenine 3-Mono-oxygenase Causes Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3271–3277.
- Liu, X.; Luo, D.; Huang, S.; Liu, S.; Zhang, B.; Wang, F.; Lu, J.; Chen, J.; Li, S. Impaired Nicotinamide Adenine Dinucleotide Biosynthesis in the Kidney of Chronic Kidney Disease. Front. Physiol. 2021, 12, 723690.
- Schefold, J.C.; Zeden, J.P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908.
- Konan, K.V.; Taylor, M.W. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 1996, 271, 19140–19145.
- Rhee, E.P.; Clish, C.B.; Ghorbani, A.; Larson, M.G.; Elmariah, S.; McCabe, E.; Yang, Q.; Cheng, S.; Pierce, K.; Deik, A.; et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 2013, 24, 1330–1338.
- Goek, O.N.; Prehn, C.; Sekula, P.; Romisch-Margl, W.; Doring, A.; Gieger, C.; Heier, M.; Koenig, W.; Wang-Sattler, R.; Illig, T.; et al. Metabolites associate with kidney function decline and incident chronic kidney disease in the general population. Nephrol. Dial. Transplant. 2013, 28, 2131–2138.
- Lee, H.; Jang, H.B.; Yoo, M.G.; Park, S.I.; Lee, H.J. Amino Acid Metabolites Associated with Chronic Kidney Disease: An Eight-Year Follow-Up Korean Epidemiology Study. Biomedicines 2020, 8, 222.
- Debnath, S.; Velagapudi, C.; Redus, L.; Thameem, F.; Kasinath, B.; Hura, C.E.; Lorenzo, C.; Abboud, H.E.; O’Connor, J.C. Tryptophan Metabolism in Patients With Chronic Kidney Disease Secondary to Type 2 Diabetes: Relationship to Inflammatory Markers. Int. J. Tryptophan Res. 2017, 10, 1178646917694600.
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943.
- Brandacher, G.; Cakar, F.; Winkler, C.; Schneeberger, S.; Obrist, P.; Bösmüller, C.; Werner-Felmayer, G.; Werner, E.R.; Bonatti, H.; Margreiter, R.; et al. Non-invasive monitoring of kidney allograft rejection through IDO metabolism evaluation. Kidney Int. 2007, 71, 60–67.
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532.
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536.
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342.e325.
- Ugur, S.; Ulu, R.; Dogukan, A.; Gurel, A.; Yigit, I.P.; Gozel, N.; Aygen, B.; Ilhan, N. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren. Fail. 2015, 37, 332–336.
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726.
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 26–33.
- Kang, Y.S.; Song, H.K.; Lee, M.H.; Ko, G.J.; Han, J.Y.; Han, S.Y.; Han, K.H.; Kim, H.K.; Cha, D.R. Visfatin is upregulated in type-2 diabetic rats and targets renal cells. Kidney Int. 2010, 78, 170–181.
- Chen, Y.; Liang, Y.; Hu, T.; Wei, R.; Cai, C.; Wang, P.; Wang, L.; Qiao, W.; Feng, L. Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-κB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp. Ther. Med. 2017, 14, 4181–4193.
- Benito-Martin, A.; Ucero, A.C.; Izquierdo, M.C.; Santamaria, B.; Picatoste, B.; Carrasco, S.; Lorenzo, O.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Endogenous NAMPT dampens chemokine expression and apoptotic responses in stressed tubular cells. Biochim. Biophys. Acta 2014, 1842, 293–303.
- Song, H.K.; Lee, M.H.; Kim, B.K.; Park, Y.G.; Ko, G.J.; Kang, Y.S.; Han, J.Y.; Han, S.Y.; Han, K.H.; Kim, H.K.; et al. Visfatin: A new player in mesangial cell physiology and diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2008, 295, F1485–F1494.
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. 2003, 63, S19–S21.
- Altmeyer, M.; Hottiger, M.O. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging 2009, 1, 458–469.
- Martin, D.R.; Lewington, A.J.; Hammerman, M.R.; Padanilam, B.J. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1834–R1840.
- O’Valle, F.; Del Moral, R.G.; Benitez Mdel, C.; Martin-Oliva, D.; Gomez-Morales, M.; Aguilar, D.; Aneiros-Fernandez, J.; Hernandez-Cortes, P.; Osuna, A.; Moreso, F.; et al. Poly polymerase-1 expression is related to cold ischemia, acute tubular necrosis, and delayed renal function in kidney transplantation. PLoS ONE 2009, 4, e7138.
- O’Valle, F.; Gomez-Morales, M.; Del Moral, R.M.; Seron, D.; Moreso, F.; Osuna, A.; Oliver, F.J.; Del Moral, R.G. Poly(ADP-ribose) polymerase expression in kidney transplantation: From alfa (alpha) to Omega (Omega). Transplant. Proc. 2007, 39, 2099–2101.
- Ogura, Y.; Kitada, M.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox. Rep. 2018, 23, 153–159.
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336.
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signal. 2018, 42, 249–258.
More