Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Huawen Huang and Version 2 by Rita Xu.
Acyl-containing organic compounds, including ketones, esters, amides, and so forth, are a huge library of widespread chemical feedstocks which play a vital role in countless fields such as pharmaceuticals, natural products, advanced materials, and fine chemicals.
- alcohols
- acyl
- acylation
1. Introduction
The classical routes to incorporate acyl group into aromatic compounds mainly rely on the Friedel–Crafts acylation reactions [1][2][3][4][5,6,7,8]. However, these methods often suffer poor selectivity, limited substrate scope, the requirement of stoichiometric moisture-sensitive Lewis acid and corrosive acyl chloride or anhydride, as well as concurrent waste production. Therefore, the development of effective and general methodologies for the selective incorporation of acyl groups into target structures has become a prevalent topic in organic synthetic chemistry. In recent years, transition-metal-catalyzed acylation reactions have attracted intensive attention and provided an efficient and straightforward synthetic route to access various acylated molecules in both academic and industrial areas [5][6][7][9,10,11]. Concomitantly, a number of simple and mild acylating reagents, such as aldehydes, carboxylic acids, esters, amides, etc., have been well explored and smoothly applied in multiple acylation transformations. Despite high efficiency, the disadvantages of these reagents should also be discussed. For instance, aldehydes are usually expensive and reactive. They are often prone to decomposing and being distilled or recrystallized prior to use in catalytic reactions. Moreover, a range of aldehydes are not naturally abundant or commercially available.
On the other hand, it is well known that alcohols are cheap, stable, low-toxic, naturally abundant, and commercially available. Thus, they are commonly used in the cross-coupling reactions [8][12], especially as oxygen-centered nucleophiles to synthesize ethers [9][10][13,14]. However, they are traditionally less used to serve as carbon-centered coupling partners. Considering the importance of these plentiful and economic chemicals in organic synthesis, alcohols can potentially act as ideal acyl precursors compared with the above reagents. Generally, the metal species react with the alcohol to yield the key alkoxide intermediate, followed by β-hydride elimination and reductive elimination to afford the final products and complete the catalytic cycle. With these strategies, a variety of alcohol acylation reactions can be found to afford a sustainable dehydrogenation manner for the construction of versatile molecules.
2. C-C Bond Construction
Acylation is a class of crucial C-C bond forming reaction which is widely involved in organic chemistry. One of its major applications is the formation of ketones, which are fundamental scaffolds found in various pharmaceuticals, agrochemicals, and natural products [11][12][13][14][17,18,19,20]. Typically, the generation of these valuable compounds relies on the addition of Grignard reagents to aldehyde, which is the most used reaction to form C−C bonds in the organic and medicinal chemistry. However, aldehyde is usually generated by the oxidation of alcohol precursor. Moreover, an additional step of oxidation is needed if ketone is the target product [15][16][21,22].
In 2019, Newman established a Ni-catalyzed cross-coupling of primary alcohols and organotriflates (Scheme 1) [17][23]. In this novel procedure, a variety of ketone frameworks were synthesized by Ni-catalyzed oxidative coupling without limitations on the substitution patterns. In addition, this method was also highly practical to the late-stage modification of bioactive molecules. Mechanistically, the use of acetone and a slight excess of triflate as terminal hydrogen acceptors was pivotal for the oxidative transformation. The combination of three distinct catalytic cycles, which contained two competitive oxidation processes and a Ni-catalyzed carbonyl Heck-type pathway, was proposed.
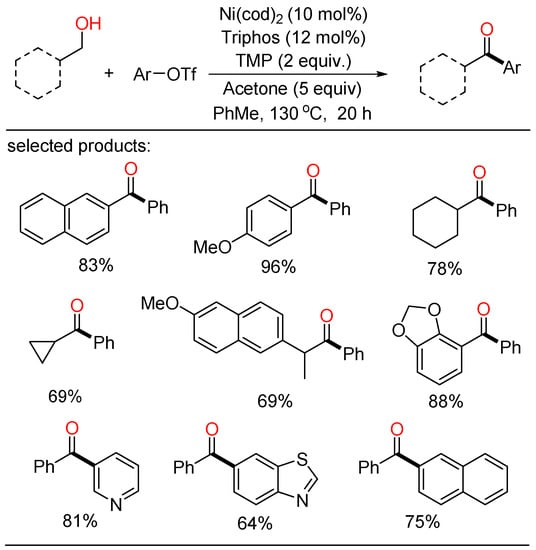
Scheme 1. Ni-catalyzed cross-coupling of primary alcohols and organotriflates.
Soon after, Satyanarayana reported palladium-catalyzed direct oxidative acylation of iodoarenes with primary alcohols, leading to the formation of various ketones (Scheme 2) [18][24]. In addition, this strategy was also applied to the synthesis of natural as well as pharmaceutical products. However, a large amount of tert-butyl hydroperoxide (TBHP) as the oxidizing agent was essential for this kind of oxidative coupling.
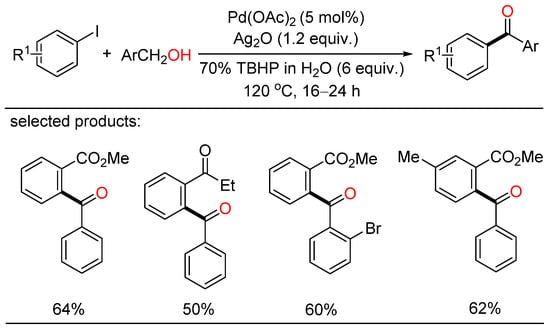
Scheme 2. Palladium-catalyzed direct oxidative acylation of iodoarenes with primary alcohols.
In 2021, Rong developed a redox-neutral cross-coupling approach for the construction of C-C bond with alkenyl primary alcohols and aryltriflates, providing a range of desired ketone products with good yields and broad functional group tolerance (Scheme 3) [19][25]. This method also afforded more functionalized ketones via the nickel/Triphos catalytic system without the addition of an external oxidant or reductant. A plausible catalytic cycle including isomerization of an alkenyl primary alcohol and a coupling process for the formation of a new C-C bond was given.
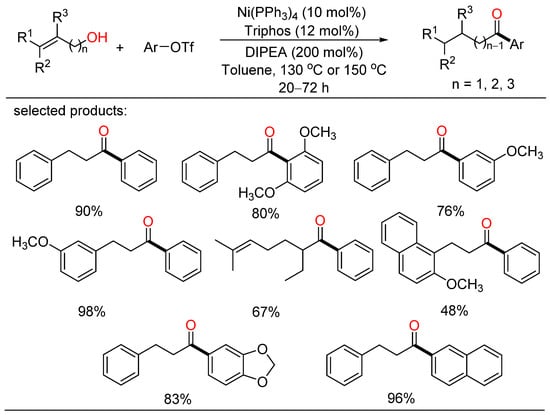
Scheme 3. Redox-neutral cross-coupling of alkenyl primary alcohols and aryltriflates.
From a synthetic point of view, the direct conversion of C-H bonds into C-C bonds can potentially result in a more efficient synthesis with a reduced number of synthetic operations and thus be a more straightforward alternative in ketone synthesis.
According to this viewpoint, Deng described a palladium-catalyzed regio-selective acylation of aryl C-H bonds with the assistance of directing group (Scheme 4) [20][26]. Several benzylic and aliphatic alcohols were chosen as the economic acyl surrogates and were oxidized in situ in the presence of peroxide. Consequently, a series of aryl ketones were gained with good yield and high regioselectivity.
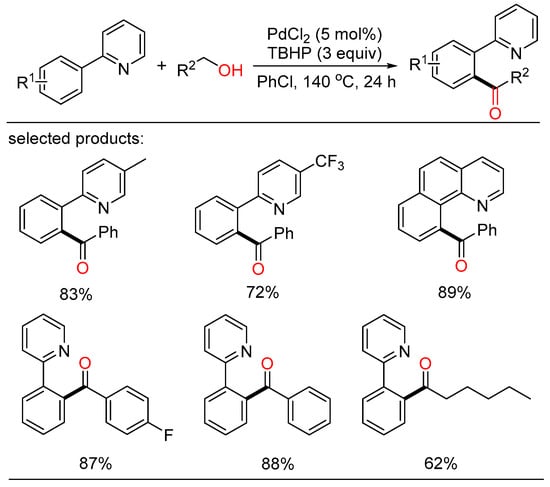
Scheme 4. Palladium-catalyzed directing acylation of aryl C-H bonds.
Compared to the above protocols which need a stoichiometric of peroxide as the oxidant, redox-triggered synthetic strategies have aroused much attention for their ability to expeditiously achieve complex ketones from simple alcohols, leading to a higher economical step to the classical synthetic routes.
In 2020, Shi demonstrated a selective NHC/Ni-catalyzed hydroacylation of alkynes with alcohols for the preparation of ketones (Scheme 5) [21][27]. A range of branched ketones were readily synthesized from various benzylic and aliphatic alcohols and internal alkynes in one step without the need for any oxidative or reductive additives.
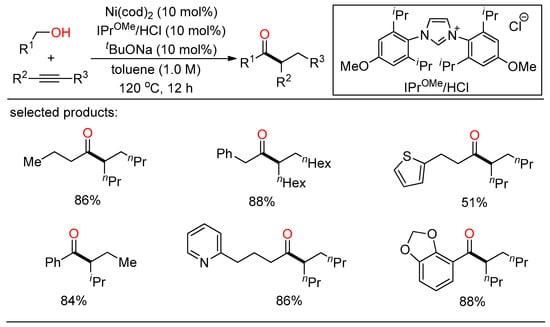
Scheme 5. NHC/Ni-catalyzed hydroacylation of alkynes with alcohols.
After a short while, Shu and Xia independently reported redox-triggered metal-catalyzed cross-coupling reactions from readily available alcohols and olefins. In Shu’s work, a variety of α-monoarylated ketones were selectively obtained by Ni-catalyzed dehydrogenative cross-coupling reaction cascade between alcohols and olefins (Scheme 6) [22][28]. This operationally simple procedure featured the utilization of commercially available and naturally abundant starting, allowing for the direct construction of monoarylated ketones with good yield, exclusive selectivity, and wide functional group tolerance. The nickel catalysts played a twofold role in two catalytic cycles. One cycle is for the oxidative dehydrogenation of alcohols to aldehydes, the other is for regioselective hydroacylation of aldehydes with olefins. On the other side, Xia disclosed a new method for the remote C(sp3)-H functionalization of olefins with alcohols (Scheme 7) [23][29]. The redox strategy was realized via a Ru-catalyzed α-acylation of olefins bearing a carbonyl group with primary alcohols, thus giving useful 1,3-dicarbonyl compounds without a base or an oxidant. Mechanistic investigations revealed that this step-economical transformation involved multiple processes, including borrowing hydrogen, hydrometallation, and metal walking.
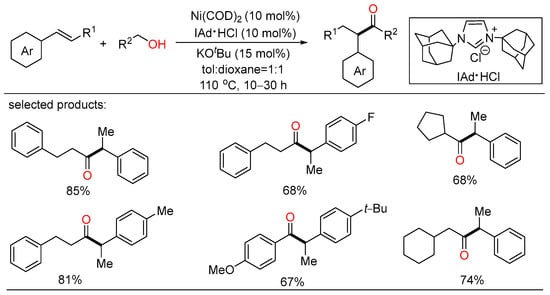
Scheme 6. Ni-catalyzed dehydrogenative cross-coupling reaction cascade between alcohols and olefins.
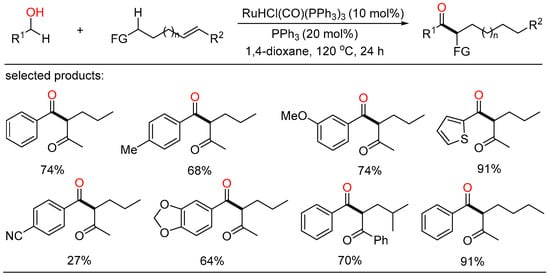
Scheme 7. Remote C(sp3)-H functionalization of olefins with alcohols.
In 2021, the first Rh-catalyzed redox-neutral conversion of primary benzylic or aliphatic alcohols with butadiene was developed by Krische (Scheme 8) [24][30]. In the protocol, various isobutyl ketones containing electronically diverse aromatic and heteroaromatic rings were successfully obtained via merged transfer hydrogenative carbonyl addition-redox isomerization. Deuterium experiments demonstrated that the rhodium alkoxide species obtained upon carbonyl addition rendered redox isomerization without dissociation of rhodium at any stage. This elegant strategy contributed an alternative way that transformed inexpensive and abundant chemical feedstocks to value-added products under simple conditions.
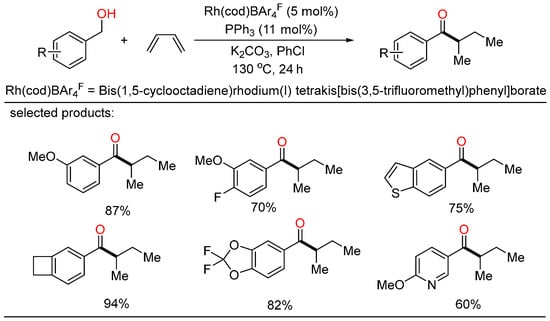
Scheme 8. Rh-catalyzed redox-neutral coupling of benzylic alcohols with butadiene.
In the meantime, Kim displayed a novel method for the synthesis of various aryl ketones from primary alcohols at room temperature (Scheme 9) [25][31]. AIn this study, alcohols were converted into acyl bromide intermediates by the action of dibromoisocyanuric acid (DBI), followed by further transformation with arene compounds in the presence of Fe2O3 via Friedel–Crafts acylation to yield the corresponding ketone products in one pot.
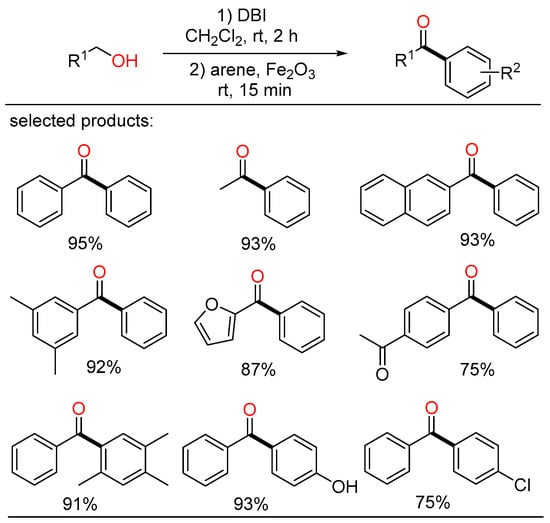
Scheme 9. Cascade coupling of alcohols with arenes.
3. C-O/C-S Bond Construction
Carboxylic acids and esters are prominent structural backbones that are widely existed in the synthesis of chemicals, drugs, materials, and polymers. Despite substantial well-established methodologies, the development of environmentally benign and cost-effective approaches for the construction of esters continues to attract great concern [26][27][32,33].
In 2010, Beller reported palladium-catalyzed oxidative esterifications of primary alcohols using dioxygen as benign oxidant, providing a general route for the synthesis of desired esters with high selectivity (Scheme 10) [28][34]. Both oxidative homocoupling reactions as well as cross-esterifications of benzyl alcohols with various aliphatic alcohols took place under mild conditions to give the corresponding esters with water as the only byproduct. In addition, these general green and selective oxidative conversions proceeded smoothly in the presence of simple catalytic systems without the aid of extra organic hydrogen acceptors. Simultaneously, Lei showed another Pd-catalyzed aerobic oxidative esterification of benzylic alcohols (Scheme 11) [29][35]. In this sustainable transformation, a range of aliphatic alcohols, including methanol and long-chain primary alcohols, successfully coupled with various benzylic alcohols to afford the target esters in moderate to high yields with good chemoselectivity. Both of these reaction pathways involving two Pd-catalyzed oxidation steps were outlined.
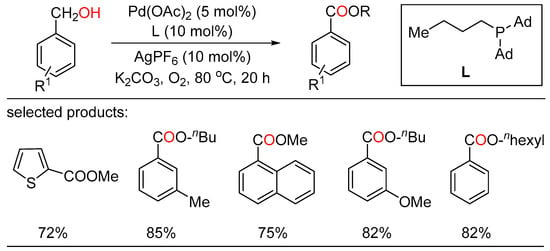
Scheme 10. Palladium-catalyzed oxidative esterifications of benzylalcohols.
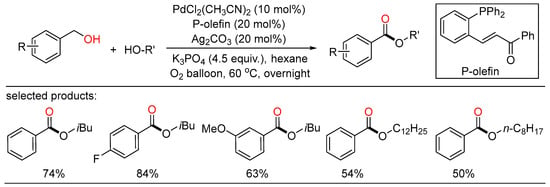
Scheme 11. Palladium-catalyzed oxidative esterifications of benzylalcohols.
In 2012, Wu developed the first Zn-catalyzed oxidation of benzyl alcohols under air at room temperature using hydrogen peroxide as benign oxidant, providing a class of methyl benzoates in moderate to excellent yields (Scheme 12) [30][36].
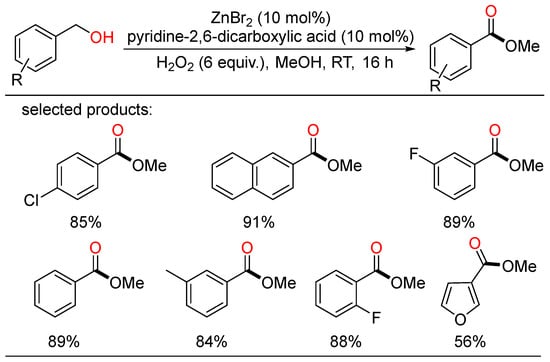
Scheme 12. Zn-catalyzed oxidative esterifications of benzyl alcohols.
In 2013, Jang discovered NHC-catalyzed oxidative coupling of allylic alcohols. Some cinnamyl alcohols were subjected to the tandem oxidation/oxidative esterification system, giving α,β-unsaturated esters in moderate to good yields with the merger of stoichiometric TEMPO as the oxidant and methanol as the additive (Scheme 13) [31][37]. The author presumed that methanol might be helpful to the deprotonation of the α-proton of cinnamyl alcohol.
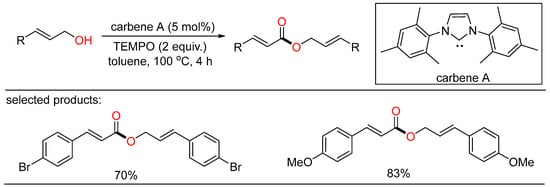
Scheme 13. NHC-catalyzed oxidative esterifications of alcohols.
Another homocoupling example of alcohols to esters was reported by Chung. They developed a robust Rh/base catalytic system that can be readily conducted to selectively yield the desired esters without ligand. A number of alcohols attached by various functionalities behaved well with nitroarene as the hydrogen acceptor (Scheme 14) [32][38].
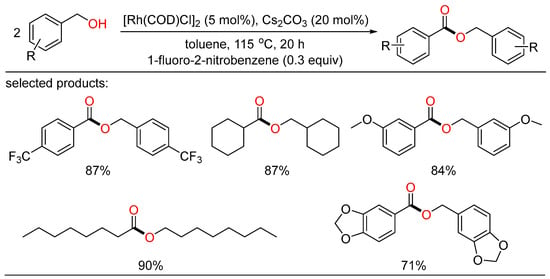
Scheme 14. Rh-catalyzed oxidative homocoupling esterifications of alcohols.
A solvent-free protocol for the copper-catalyzed selective oxidation of primary alcohols to esters was developed by Wei. Under neat conditions, homocoupling esterification and cross-esterification of benzylic alcohols with various aliphatic alcohols proceeded successfully to afford the target esters in good yields (Scheme 15) [33][39].
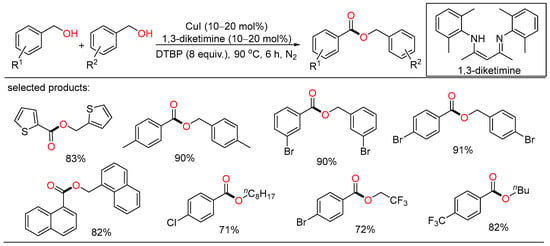
Scheme 15. Cu-catalyzed oxidative esterifications of alcohols.
In 2014, Fu and Yuan jointly reported copper-catalyzed construction of C-O bonds through oxidative coupling of benzylic alcohols with ethers in open air (Scheme 16) [34][40]. A variety of α-acyloxy ethers were garnered in good yields with TBHP as the oxidant. A TBHP-initiated, copper-promoted radical mechanism for this oxidation reaction was proposed.
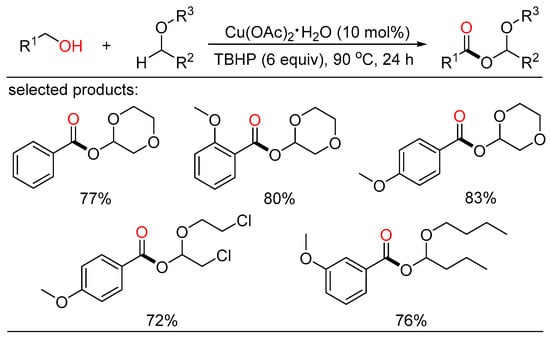
Scheme 16. Cu-catalyzed oxidative esterifications of alcohols with ethers.
Meanwhile, Maiti and Lahiri unveiled novel approaches towards selective cross- and self-oxidative esterification of a broad range of alcohols (Scheme 17) [35][41]. The cross-esterification occurred under a transition-metal-free condition with the combination of catalytic TEMPO/TBAB and Oxone® as the oxidant, while the self-esterification was accomplished by the involvement of Fe(OAc)2/pyridine-2,6-dicarboxylic acid as the active catalyst.
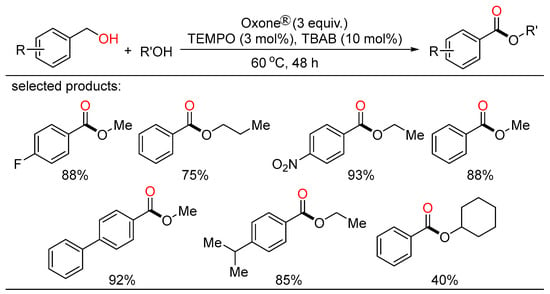
Scheme 17. Oxone-mediated oxidative esterifications of alcohols.
In 2017, Elias described I2/NaOH-catalyzed oxidation of alcohols for the preparation of carboxyl acids using water as the solvent (Scheme 18) [36][42]. A series of aryl- and heterocycle-substituted carboxyl acids were efficiently obtained with good yield under mild reaction conditions. Mechanistic studies indicated that IO2− is the most probably active catalyst which was formed and regenerated by the oxidation of IO− by TBHP. This metal-free protocol did not require chromatographic purification, featuring operational convenience, broad substrate scope, and easy scalability.
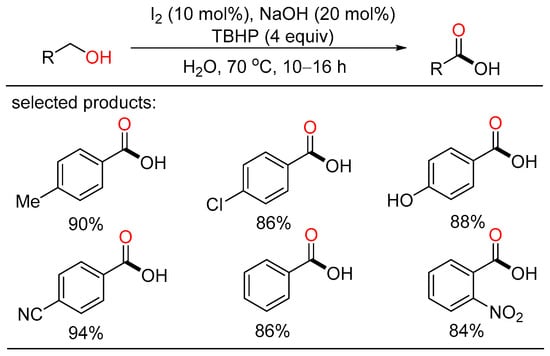
Scheme 18. I2-catalyzed oxidative carboxylation of alcohols.
Later, Wei reported a novel Anderson-type chrome-based catalyst which can render the direct transformation from alcohols to esters by H2O2 oxidation in good yields and high selectivity without extra ligands (Scheme 19) [37][43]. A variety of alcohols with different functionalities including some natural products and pharmaceutical intermediates are tolerated in this system. The catalyst can be applied to gram-scale reactions, and retain good catalytic reactivity after recycling several times, indicating potential value of the catalytic system in industrial realm.
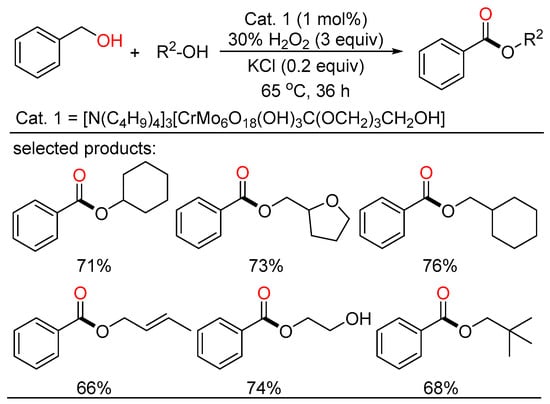
Scheme 19. Chrome-catalyzed oxidative esterifications of alcohols.
In 2022, Bolm exploited a mechanochemical procedure for the palladium-catalyzed oxidative esterification of alcohols, allowing a solvent-free synthesis of symmetrical and unsymmetrical esters in up to excellent yields after short reaction times at ambient temperature (Scheme 20) [38][44]. Manifold liquid and solid benzylic and aliphatic alcohols were easily self-esterified and cross-esterified under such mild reaction conditions. Moreover, secondary alcohols were also carried out in the cross-esterification with benzylic alcohols. This sustainable protocol enabled significant waste reduction by evaluating some metrics, providing a complement to solution-based methods.
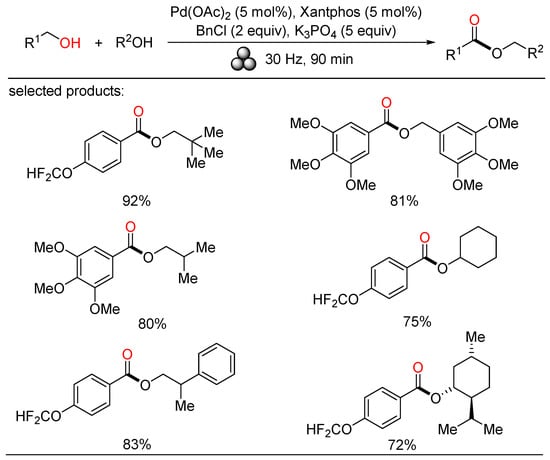
Scheme 20. Pd-catalyzed mechanochemical oxidative esterifications of alcohols.
At the same time, Yamaguchi showed a highly selective method for the selective aerobic oxidation of aliphatic primary alcohols, demonstrating the formation of oxalic acid diesters via oxidative esterification from alcohols and ethylene glycol using the CuCl/TMEDA/DMN-AZADO catalytic system (Scheme 21) [39][45]. This novel project was fulfilled under mild reaction conditions using dioxygen as the terminal oxidant and only required a stoichiometric ratio of alcohols and ethylene glycol. Detailed experiments and DFT calculations were conducted to elucidate the mechanism of the novel oxidation reactions.
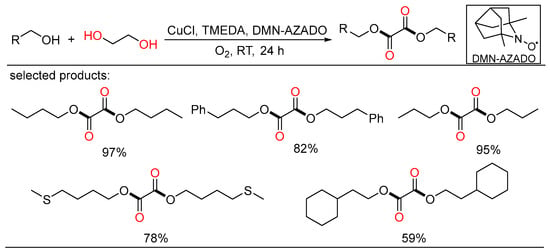
Scheme 21. Cu-catalyzed oxidative diesterifications of alcohols.
Although the oxidative coupling reactions of alcohols provide efficient patterns to acquire esters, from the viewpoint of sustainable chemistry, the acceptorless dehydrogenative coupling (ADC) reaction of alcohols would be the most promising method for ester synthesis because it appears as an atom- and step-economical process accompanied by the extrusion of gaseous hydrogen as the only byproduct (Scheme 22).
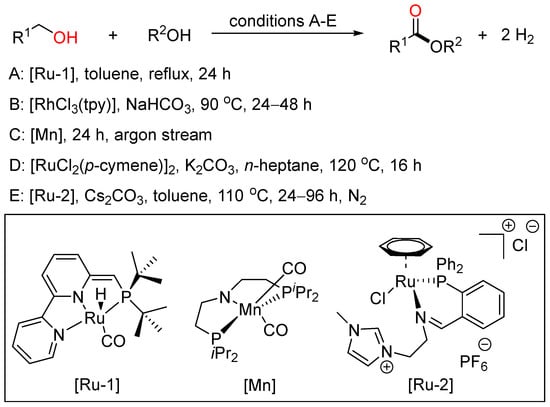
Scheme 22. Transition metal-catalyzed dehydrogenative esterifications of alcohols.
In 2012, Milstein employed a bipyridyl-based Ru(II) pincer catalyst to realize cross-dehydrogenative coupling of primary alcohols with secondary alcohols, affording esters with the liberation of dihydrogen in high yield and good selectivity under neutral conditions (A) [40][46]. In 2016, Xiao reported Rh-terpyridine catalyzed dehydrogenative cross-coupling of primary alcohols to yield esters with broad substrate scope, good functional group tolerance and high selectivity (B) [41][47]. Then, Gauvin described aliphatic pincer-supported earth-abundant manganese complexes as effective catalysts for the acceptorless dehydrogenative coupling of a wide range of alcohols to esters under base-free conditions. The reaction proceeded smoothly under neat conditions, with modest catalyst loading and releasing only dihydrogen as byproduct (C) [42][48]. Then, Zhao and Li cooperatively developed a Ru-catalyzed highly selective dehydrogenative cross-coupling of alcohols to esters using phosphine oxide containing ligands based on the aldehyde effect (D) [43][49]. Recently, Fu and Li together presented the acceptorless dehydrogenative cross-coupling of primary alcohols to access esters effected by designing a novel Ru complex as the catalyst. These two sustainable protocols were tolerated to a wide range of primary alcohols to give various unsymmetrical esters with good functional group compatibility and high selectivity (E) [44][50].
The ADC strategy was also applied in the synthesis of carboxylic acid. Bera and Verpoort reported Ru-catalyzed direct conversion of alcohols to carboxylic acids under basic conditions, respectively. In the former method, a one-pot transformation of alcohols to carboxylic acids proceeded using alkaline water (Scheme 23) [45][51]. Various primary alcohols such as benzyl alcohols, long-chain alcohols, amino alcohols, and diols were tolerated in the catalytic system. Mechanistic studies indicated two sequential processes, namely acceptorless dehydrogenation of alcohols to aldehydes, and following “aldehyde-water shift” reaction. In the latter work, Verpoort et al. developed a graceful method of acceptorless dehydrogenative coupling of alcohols with hydroxides by orchestrating a series of NHC-Ru complexes (Scheme 24) [46][52]. Then, they evaluated the effects of several substituents on the catalytic performance of the resulting NHC-Ru complexes. Gram-scale preparation of carboxylic acids was also conducted at an ultralow catalyst loading in open air.
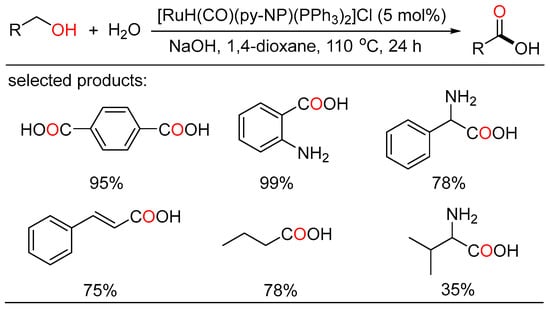
Scheme 23. Ru-catalyzed dehydrogenative carboxylation of alcohols.
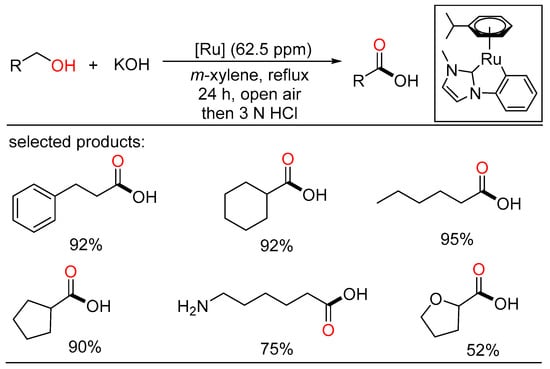
Scheme 24. Ru-catalyzed dehydrogenative carboxylation of alcohols.
As a family of activated carboxylic acid derivatives, thioesters are very versatile building blocks in the fields of chemistry and biology. Based on the importance of these compounds, Zhu disclosed a tetraethylammonium bromide (TEAB)-catalyzed oxidative coupling of alcohols with thiophenols or disulfides (Scheme 25) [47][53]. This metal-free protocol offered a convenient route to the formation of a wide range of thioesters with broad substrate scope in high yields. Mechanistically, cross-coupling of the acyl- and sulfur radicals generated in the oxidizing environment resulted in the formation of the corresponding thioesters.
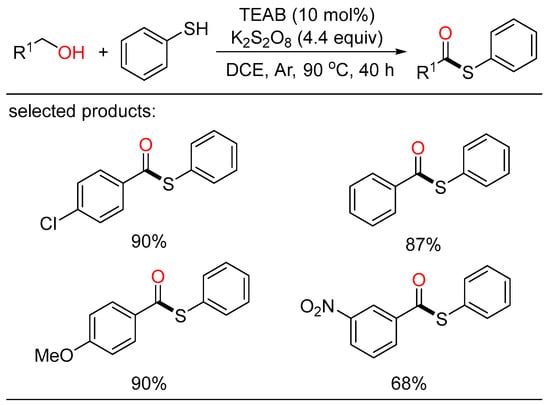
Scheme 25. TEAB-catalyzed oxidative coupling of alcohols with thiophenols.