Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Miao Liu and Version 3 by Conner Chen.
The PROteolysis TArgeting Chimeras (PROTACs) is an innovative technique for the selective degradation of target proteins via the ubiquitin–proteasome system. Compared with traditional protein inhibitor drugs, PROTACs exhibit advantages in the efficacy and selectivity of and in overcoming drug resistance in cancer therapy, providing new insights into the discovery of anti-cancer drugs.
- PROTACs
- cancer therapy
- targeted protein degradation
1. Introduction
Despite the continuous development of new treatment methods, malignant tumors still seriously endanger human health [1]. Compared with traditional chemoradiotherapy and surgical treatments, individually targeted approaches and immunotherapy have been large breakthroughs for the treatment of malignant tumors over the past 20 years. Currently, they are highly active areas of research. At present, the molecularly targeted drugs that have been used in clinical treatments are mainly small molecule inhibitors and monoclonal antibodies, which bind to the active sites of the target proteins as competitive antagonist ligands to hinder the target protein’s ability to bind to their downstream targets. However, drug resistance may occur due to changes in the binding sites caused by gene mutations or alterations of the conformation of the target protein. At present, though traditional targets (such as kinases and G protein-coupled receptors) are still the main research focus, the landscape of the drug target field is changing, slowly shifting from traditional drug targets to more challenging “undruggable” targets. These targets typically include proteins that have no enzymatic function which account for about 80% of the total human proteins.
PROTACs, PROteolysis-TArgeting Chimeras, are heterobifunctional small molecule compounds, which consists of a ligand for the target protein, a linker, and a ligand to recruit E3 ligase. It can induce the degradation of the target proteins, including the “undruggable” targets. It is a new strategy for the research and development of novel small molecule drugs, and it can address the dilemma of the known problems of using targeted drugs [2]. At present, several PROTAC drugs have undergone or are undergoing clinical trials, including ARV-110 [3] which targets the androgen receptor in prostate cancer, ARV-471 [4], targeting the estrogen receptor in breast cancer, and FHD-609 which targets BRD9 in Synovial sarcoma.
To date, PROTACs have been successfully applied in the degradation of many protein targets. However, there are still some limitations to PROTACs that need to be overcome through future research. Due to their special structures, the PROTACs usually have a high molecular weight and a poor performance in pharmacokinetics. Although many PROTACs have been reported to exhibit activities in vivo, and they are clinically studied now due to their special structures, the PROTACs usually have a high molecular weight and a poor performance in pharmacokinetics and druggability studies [5][87]. The toxicity and side effects of PROTACs are also a large concern in terms of their application in the clinic. On one hand, unlike the traditional inhibitors which only inhibit the activity, but do not affect the expression of the target proteins, the PROTACs induce the degradation of the target protein, by which drug resistance may be prevented but a higher irreversible toxicity could be generated [6][88]. On the other hand, the off-target effect of the PROTACs may induce severe consequences for affecting the physiological activities of the normal cells or organs. Therefore, the close monitoring and tracking of the toxicological effects are needed as for the clinical studies and in the late stages of the drug development of PROTACs [7][89].
2. Structure Character and Mechanism of PROTACs
The main pathway for protein degradation in the cells is ubiquitin–proteasome system-mediated degradation. E3 ubiquitin ligase recognizes and ubiquitinates the target proteins, and the ubiquitinated target proteins enter the 26S proteasome for degradation [8][5]. On the basis of the biological rationale of ubiquitination-mediated degradation, proteolytic targeting chimera (PROTAC) has been developed. Many targets that cannot be regulated by small molecules or antibodies can be affected by PROTAC technology. Unlike the traditional role of small molecule drugs which aim to block or inhibit the function of proteins, the novelty and usefulness of PROTAC is its ability to send any target proteins into the proteasome for degradation [9][6]. As a heterobifunctional molecule, PROTAC is composed of three parts (Figure 1): a target protein ligand, an E3 ligase ligand, and a linker that connect the two ligands to form a “three-body” polymer, the target protein ligand-linker-E3 ligand. It uses the ubiquitin–proteasome pathway to specifically degrade the target protein by bringing the target protein closer to the E3 ubiquitin ligase in the cell [3]. As shown in Figure 1, PROTAC-induced POI degradation includes four major steps: Firstly, PROTAC binds to the POI and E3 ligase to form a ternary complex (POI-PROTAC-E3 ligase). Secondly, E3 ligase recruits E2 ligase and mediates the transfer of ubiquitin (Ub) from the E2 to lysine residues on the POI. Thirdly, the ubiquitination occurs several times to form multi-Ub chains on the surface of the POI. Finally, the ternary complex dissociates, and the polyubiquitinated POI is degraded by the 26S proteasome. The dissociated PROTAC can then participate in a new round of degradation [10][7].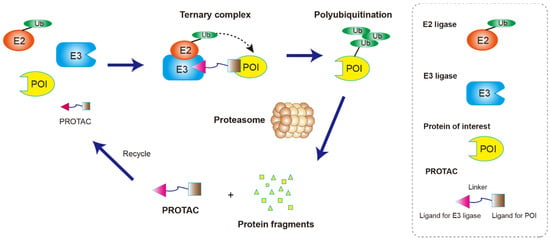
Figure 1. Illustration of the PROTAC mechanism. PROTACs are composed of a ligand that binds to a protein of interest (POI) which is tethered to another ligand that binds to the ubiquitin ligase enzyme through a short chemical linker. A POI-PROTAC-E3 ligase ternary complex is formed which leads to tagging of the protein of interest with ubiquitin groups. The proteasome then recognizes the polyubiquitination signal and mediates the degradation of the target protein. The PROTAC molecule can be recycled for another degradation cycle.