Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Roshanak Sharafieh.
Cellular senescence has gained increasing attention in the field of aging research. Senescent cells have been implicated in biological aging processes, tumorigenesis, development, and wound repair amongst other processes and pathologies. Recent findings reveal that senescent cells can both promote and inhibit cutaneous wound healing processes.
- wound healing
- acute cutaneous wounds
- chronic cutaneous wounds
1. Cellular Senescence
Cellular senescence is a cell fate defined by a permanent arrest from the cell cycle [2,3,4,6][1][2][3][4]. This cell state was first described in the 1960’s in human diploid cells that had undergone serial passages to the point of non-division [7][5]. Beyond replicative stress, senescence is also elicited by other stressors to the cell such as metabolic, oxidative, and oncogenic stress [2,3,8][1][2][6]. Additionally, such stressors can provoke telomere shortening, DNA damage pathway activation, mitochondrial dysfunction, and oncogene induction [2,3,8][1][2][6]. Morphologically, senescent cells are larger than non-senescent counterparts and demonstrate enhanced granularity associated with their altered metabolism and increased lysosomal content [2,3,8][1][2][6]. At the transcriptomic level, there are also widespread changes in gene expression within senescent cells which have been reviewed previously [4,9][3][7]. A great deal of interest in the field has been inspired by findings linking cellular senescence to a number of age-related pathologies as well as biological aging itself [2,4,8][1][3][6]. In particular, there is evidence that the removal of senescent cells in an organism may lead to an increase in health span and lifespan, and conversely that the addition of senescent cells to a previously healthy organism may increase overall morbidity and mortality [2,10][1][8].
2. Senescence Characteristics
Senescent cells are heterogeneous, and currently there is no single marker that can define a cell as senescent [2,8][1][6]. Furthermore, these cells produce a typically pro-inflammatory secretome referred to as the senescence associated secretory phenotype (SASP), which possesses corresponding complexity [2,4,8,11,12][1][3][6][9][10]. The SASP is responsible for paracrine and endocrine signaling via chemokines, cytokines, growth factors, and proteases that are all released from the senescent cells and can signal other neighboring cells into a state of senescence termed “paracrine senescence” [2,13][1][11].
There are a number of ways that senescent cells can be detected experimentally. Several of these techniques rely upon increased organelle dysfunction present in senescent cells. Mitochondria accumulate in senescent cells due to their altered metabolism [2,9][1][7]. Reactive oxygen species (ROS) production, redox state, mitochondrial function, and mitochondrial biogenesis assays can peripherally inform investigators of senescent cell burden [2][1]. Lysosome accumulation is another common indicator of cellular senescence [2,9][1][7]. Two markers of senescence in the lysosome include lysosomal senescence-associated beta-galactosidase (SA beta-Gal) and lipofuscin [2,9][1][7]. Both of these markers can be measured using an activity assay, which is a simple test to profile senescent cells [2,9][1][7]. Along with mitochondrial and lysosomal overabundance, the nucleus is also affected by the activation of a senescent state [2][1]. DNA damage response pathways lead to activation of the cyclin dependent kinase inhibitors p16 and p21 and subsequent cell cycle arrest. Other nuclear markers include phosphorylation of histone H2AX and the related telomere associated foci [2,9][1][7]. DNA Segments with Chromatin Alterations Reinforcing Senescence, or DNA-SCARS, are another type of DNA damage found within senescent cells referring to persistent damage of DNA marked by DNA damage response proteins [2,9,14][1][7][12]. DNA-SCARS are also called senescence-associated DNA damage foci, and are associated with growth arrest and interleukin 6 (IL-6) secretion in senescent cells [14][12]. RNA sequencing of senescent cells reveals that SASP-associated gene expression presents an additional layer of heterogeneity [8][6]. Sequencing data reveal differential gene expression of senescent cells dependent on stressor type, cell type, and duration of senescent state [15][13]. A new algorithmic technique that involves two phases of testing is under development for defining a senescent state via detecting multiple hallmark characteristics [16][14]. Phase one of the assessment validates the cells are in a senescent state by testing markers such as: SA-B-Gal or lipofuscin, p16, p21, lamin B1, and SASP proteins [16][14]. After phase one defines the presence of senescence, phase two allows for characterization of the senescence subtype by testing for transcriptomic features, for example pro-inflammatory SASP transcripts, as well as secreted proteins [16][14].
The heterogeneity of cellular senescence poses a distinct challenge because there is not one common defined marker that is present in every senescent cell [2,8,17][1][6][15]. There has been some success in identifying several common markers in senescent cells including activation of the p16/retinoblastoma and the p21/p53 signaling pathway, as mentioned previously [2,8][1][6]. Due to this, p16 and p21 gene expression and protein levels are common indicators that a cell is in a senescent state [2,4,18,19,20,21][1][3][16][17][18][19]. Other markers include p19, uPAR, and glycoprotein non-metastatic melanoma protein B (GPNMB) [8][6]. While both p16 and p21 expression are common markers leveraged by the field, it is important to note their limitations. In particular, these proteins may be transiently upregulated in diverse biologic contexts. Examples of their “non-senescent” upregulation include p21′s roles in embryologic development and hair growth cycles, or the presence of p16 in embryonic tissues and basic macrophage physiology [8,17,22,23,24,25][6][15][20][21][22][23]. Additionally, the accumulation kinetics of these proteins vary during the initiation of a senescent state [26][24]. A study using a radiation-induced osteoporosis model suggests that p21 is upregulated when the cell first becomes senescent, while later p16 activation maintains the senescent state [3,26][2][24]. However, this work also suggested that the individual roles of p21 and p16 appear to be independent of one another in the context of the model system used [26][24]. Bulk RNA sequencing data from human fibroblast and mouse fibroblasts shows a difference in transcriptomic signatures and SASP products based on senescence inducer, cell type, and stage of senescence [8,15][6][13]. To further validate the heterogeneity of senescent cells, a study revealed that different senescent cell inducers (oncogenic stress, replicative stress, IR-induced, and Dox-induced) applied to the same cell line evoke differential RNA expression supporting that senescence is a diverse state depending on cell lineage and stress type [27][25]. Furthermore, this heterogeneity is recapitulated at the single cell resolution as demonstrated by the variation of transcriptomic signatures of senescent cells found within individual cultures of human fibroblasts subjected to identical culture conditions [8,28][6][26]. As cellular senescence may be elicited by different stressors across environmental contexts, multiple markers should be tested when assessing SASP production in senescent cells [8,17][6][15].
3. Cutaneous Wound Healing
One of the primary functions of skin is to provide an environmental barrier, which creates a sterile environment for the underlying cells and connective tissue. Central to maintaining this barrier is the balance of colonization between commensal (non-pathogenic) microbes (e.g., Staphylococcus epidermidis), and pathogenic microbes (e.g., Staphylococcus aureus) [29,30,31,32][27][28][29][30]. Commensal microbe colonization of skin maintains skin barrier function by suppressing pathogenic microbe colonization and promoting low-level/baseline innate immunity on local skin to monitor and quickly respond to any barrier breaches (Figure 1). Alternatively, colonization of skin by pathogenic microbes, destroys the architecture and function of the skin, by releasing microbial toxins and proteases that directly breakdown key skin barriers (e.g., keratinocytes and connective tissue), as well as inducing acute and chronic inflammation, which in turn, destroys the cells and matrix in the skin (Figure 1). Additionally, mechanical injury (e.g., abrasions, cuts or burns) to the skin also breach the skin barrier and promote both pathogenic microbe colonization, as well as tissue destructive acute and chronic inflammation, all of which can compromise wound healing [32,33,34,35][30][31][32][33]. Thus, controlling skin microbe colonization, mechanical injury of skin, and related tissue reactions (inflammation and wound healing) is key to both maintaining and re-establishing effective skin barrier function. Since inflammation and wound healing are central for maintaining and re-establishing skin barrier function, it is important to understand these processes, as well as the impact of aging on their function in both normal and aging skin [17,29,31,32,33,36,37,38,39][15][27][29][30][31][34][35][36][37].
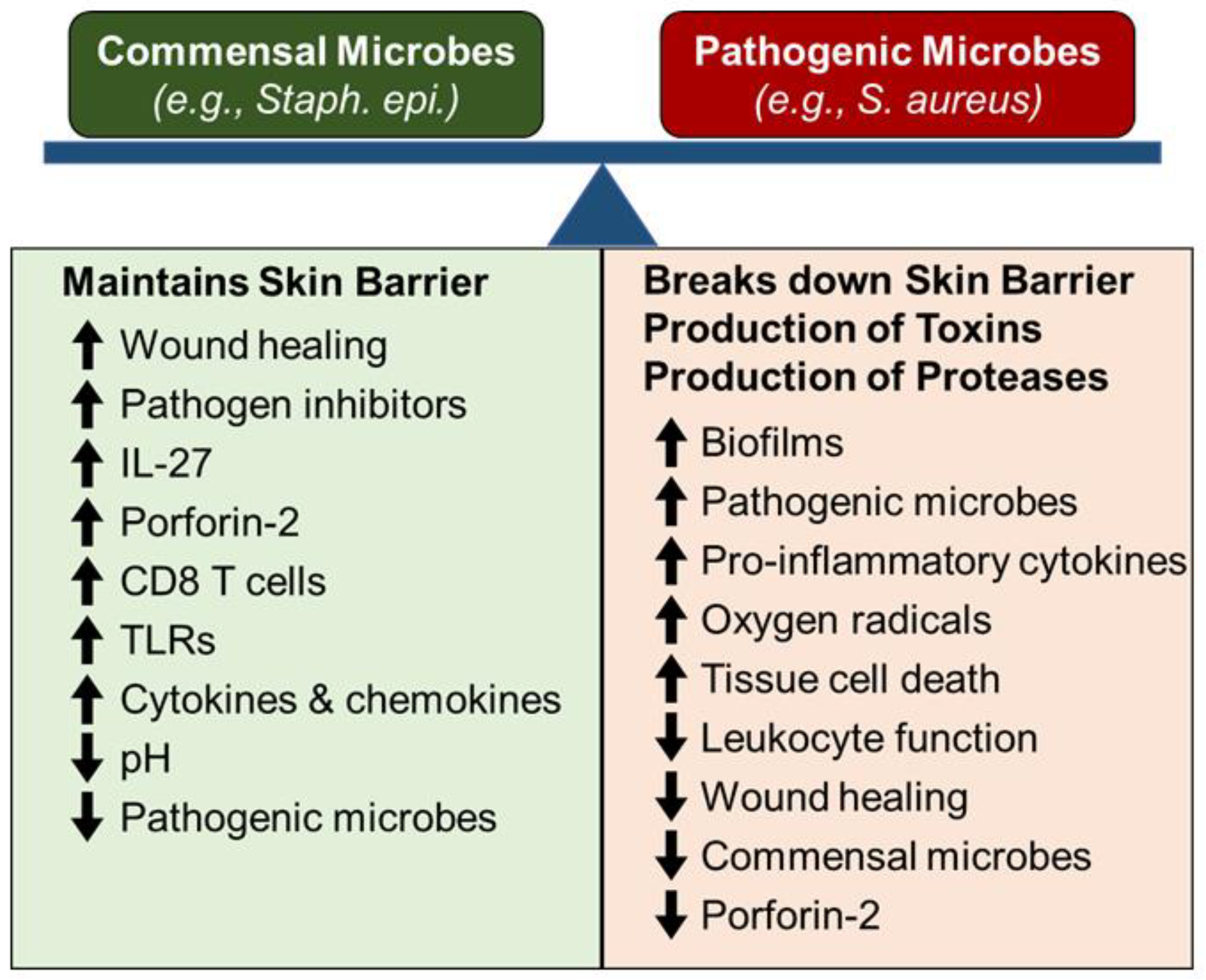
Figure 1.
The skin microbiome balance and skin wound healing.
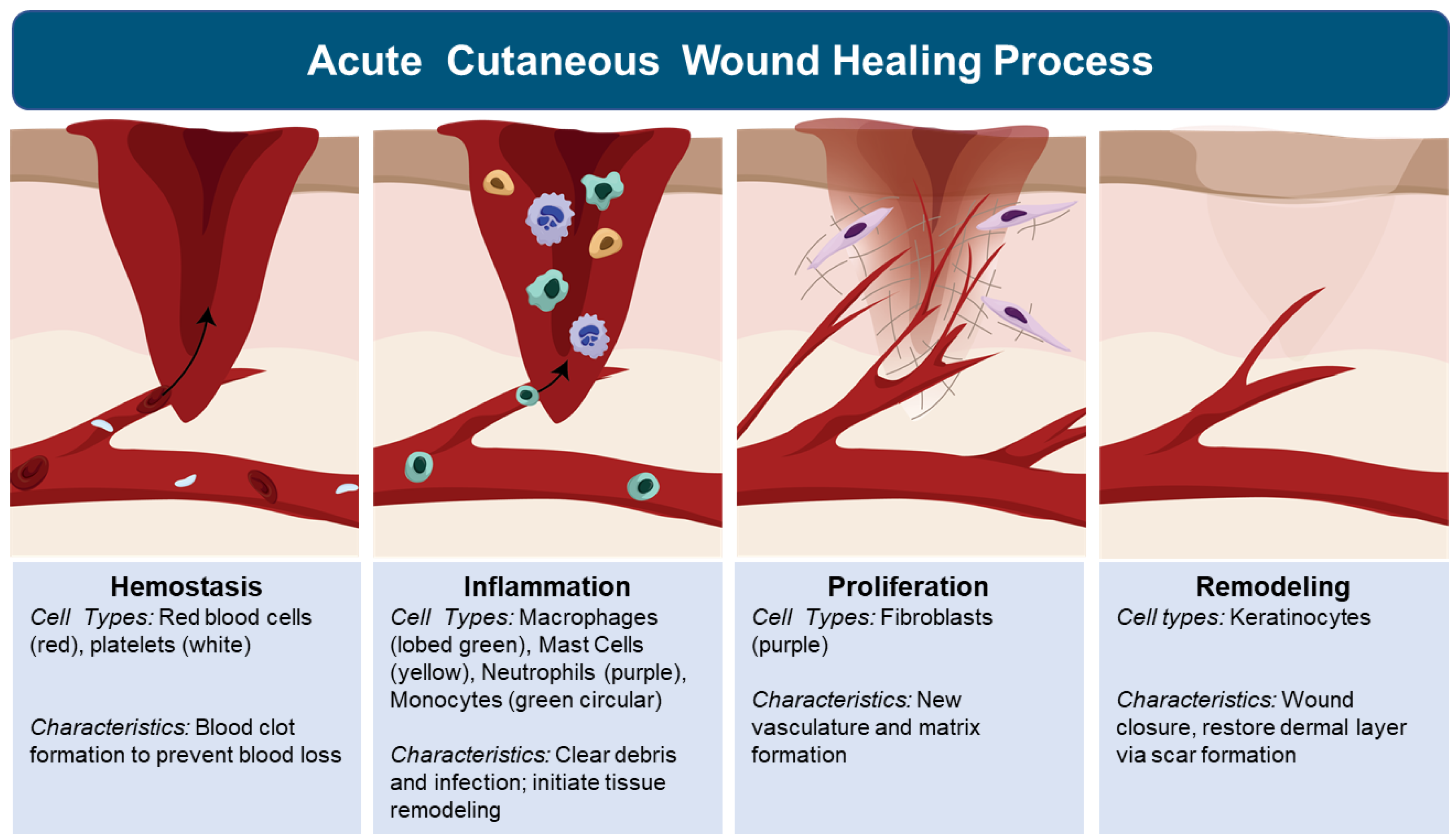
Tissue response to injury, regardless of organ, begins with inflammation. The inflammatory response is triggered to protect the body by localizing and eliminating the damaged tissue to allow the body to heal. The inflammatory phase of tissue response to injury, is activated by skin mast cell release of vaso-active amines with increased vaso-permeability of blood plasma (edema) within the injured tissue [6,33,35,39][4][31][33][37]. This initial wave of fluids to injured tissue provides immediate clot formation (Figure 2). Besides preventing further bleeding, the clot serves as a temporary tissue matrix to stabilize the injury and presents a barrier to the environment, including pathogenic microbes [6,33,35,39][4][31][33][37]. This movement of fluids is followed by a surge of leukocytes, which include neutrophils, followed by macrophages and lymphocyte infiltration (Figure 2) [6,33,35,39,40][4][31][33][37][38]. Neutrophils function as the first line of defense, killing pathogens and clearing cellular debris [40][38]. Monocytes circulate in the bloodstream and differentiate into macrophages at the wound site where they phagocytose dead neutrophils, pathogens, and tissue debris and release pro-inflammatory cytokines (e.g., IL-1, IL-6, IL-8, and TNF-α) that can sustain or further amplify the inflammatory response to resolution or to chronic inflammation leading to tissue destruction [41][39]. Central to determining these outcomes are macrophage subpopulations, including M1 and M2 macrophages [6,33,34,35,37,39][4][31][32][33][35][37]. M1 macrophages promote inflammation which clears injurious agents but can also destroy tissue structure and function. M2 macrophages promote wound healing by stimulating keratinocyte, fibroblast, and endothelial cell proliferation and migration, which assists wound transition to the proliferation phase [41][39]. During the proliferation phase, fibroblasts weave collagen fibers to repair tissue structure, and provide matrices for cell migration to occur (e.g., endothelial cell migration needed to form new vasculature to sustain the healing wound tissue) (Figure 2) [40][38]. The formation of granulated tissue signals an important transition, in which keratinocytes migrate across the newly formed matrix to close the wound [40][38]. Although the migration of keratinocytes does restore the epidermis layer of skin, the architecture and function of the dermis is lost with scar formation (Figure 2) [6,33,35,39][4][31][33][37]. This cascade of tissue responses is central to understanding and controlling cutaneous inflammation and wound healing. Although there is significant information that exists related to inflammation and wound healing in the skin of healthy individuals, the impact of aging and roles senescent cells play on inflammation and wound healing, is only beginning to be unraveled.
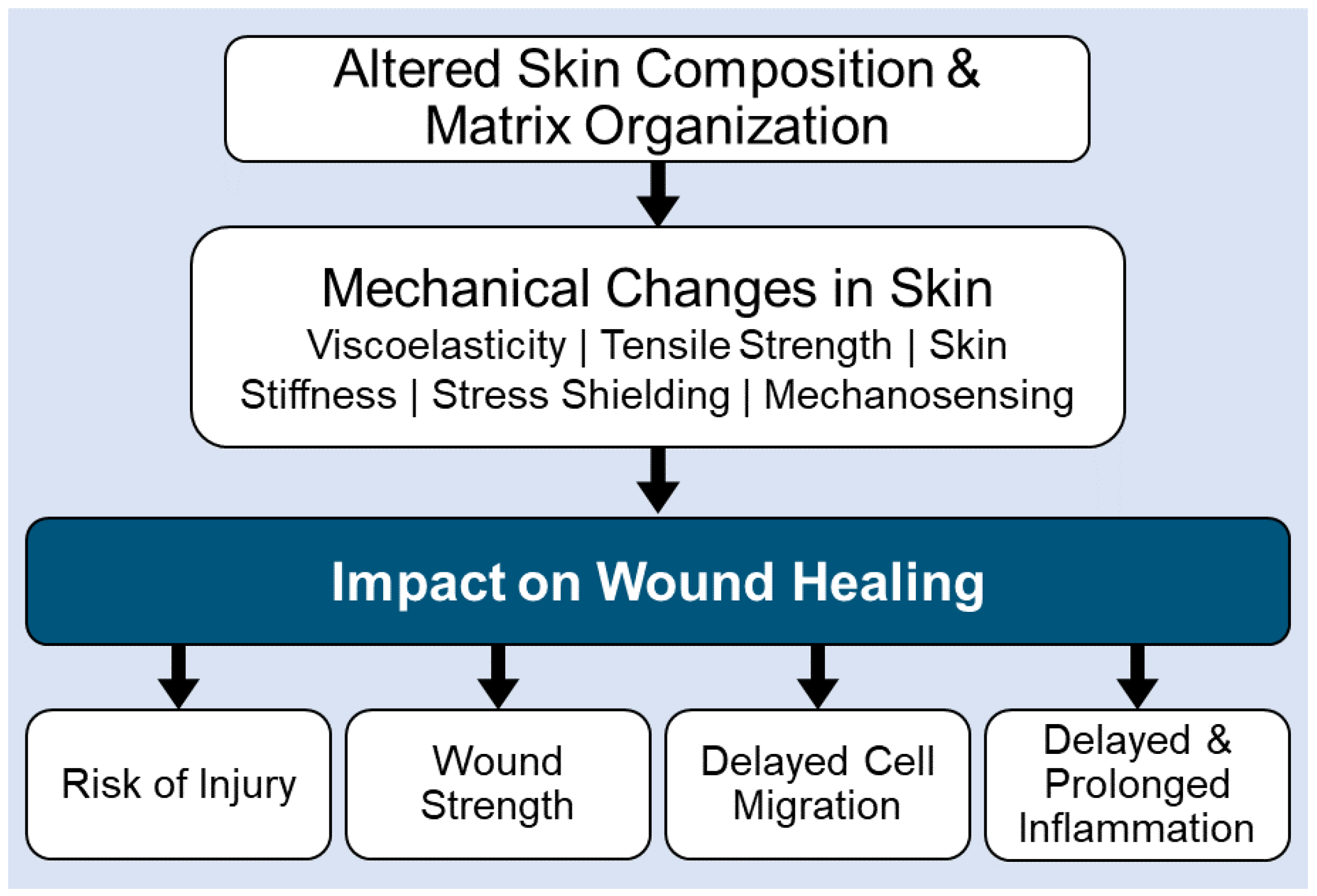
Aging can impact the skin microbiome (e.g., commensal vs. pathogenic microbe colonization) by changes to the skins’ structure and function. For example, alterations in hormonal, metabolic, and/or immune systems, increased wrinkle formation, decreased elasticity, defective wound healing, decline in the production of sebum, and decreased water content, can alter microbe colonization patterns in the skin [6,17,29,30,31,32,34,35,36,37,38,39][4][15][27][28][29][30][32][33][34][35][36][37]. Age-related loss of effective immunity in the skin, promotes the colonization of pathogenic microbes, which in turn promote chronic inflammation and excessive tissue destruction in the skin. Unfortunately, aging also negatively impacts the cells that are critical to tissue remodeling by altering skin architecture and function (Figure 3). This includes the accumulation of senescent tissue cells (keratinocytes and fibroblasts), decreased cell migration capabilities, modified extracellular matrix remodeling with diminished fiber density and increased matrix fragmentation, loss of elastin networks, and glycosaminoglycan alterations. These age-related changes in the skin architecture, alter viscoelasticity, tensile strength, skin stiffness, stress shielding, and mechanosensing [6,17,29,30[4][15][27][28][29][30][32][33][34][35][36][37],31,32,34,35,36,37,38,39], all of which lead to defective wound healing in the aging populations (Figure 3).
4. Practical Applications to Target Senescent Cells
Senescent cells can be pharmacologically targeted using senolytics. This putative drug class is designed to specifically eliminate senescent cell populations while sparing non-senescent counterparts, which has been shown to delay the aging process in murine models [61][41]. The discovery of senolytics has increased the potential for treating multiple age associated diseases simultaneously. Senescent cell clearance via senolytics may be an effective strategy to promote closure of chronic wounds, similar to their therapeutic potential in other chronic diseases. There are several subclasses of therapeutic agents that have been repurposed or used as senolytics. This includes BCL-2 inhibitors, flavonoids, and metformin amongst others that have been thoroughly reviewed [62,63,64,65,66,67,68,69][42][43][44][45][46][47][48][49].
Members of the BCL-2 protein family can act to either inhibit apoptosis in cells, thus promoting cell survival, or serve as pro-apoptotic agents [62][42]. In a typical cell, these BCL-2 partners will be balanced [62][42]. When anti-apoptotic pathway members predominate, the cell is arrested at the G0 phase of the cell cycle [62][42]. A BCL-2 inhibitory senolytic drug, Navitoclax, promotes apoptosis in a wide range of senescent cells via sequestration of anti-apoptotic BCL-2 family members, though it does not eliminate all senescent cells [13,70][11][50]. Navitoclax has limited range of impact on senescent cells due to heterogeneity in senescent cell populations which can rely on different anti-apoptotic pathways for survival [13,71][11][51]. Additionally, Navitoclax is associated with toxic side effects (e.g., thrombocytopenia and neutropenia) [13][11]. Navitoclax has not been applied to the treatment of chronic wounds yet, although another senolytics agent, UBX0101, has been used [72,73][52][53]. Studies have shown that the injection of UBX0101, into aged mice cleared senescent cells and reduced symptoms of osteoarthritis [72,73][52][53]. A clinical trial adapted UBX0101 treatment for osteoarthritis into humans but, was paused in phase II due to inability to outperform the placebo group [2][1].
The use of flavonoids is a common strategy to clear senescent cells from murine models [10,26][8][24]. Flavonoids are natural phenolic structures found in plants that are associated with beneficial impacts on health [63][43]. Flavonoids are known to be antioxidative, anti-inflammatory, anti-mutagenic, and anti-carcinogenic with chemical properties that enhance key enzymatic functions and pathways [63][43]. Two common flavonoids used in senescence research are Quercetin (Q) and Fisetin (F) [2,13][1][11]. Although it exerts senolytic activity when used alone, the flavonoid Quercetin is typically coupled with Dasatinib, a SRC/tyrosine kinase inhibitory chemotherapeutic agent [13][11]. This combination therapy is used to exploit synergistic effects that target multiple senescent cell associated anti-apoptotic pathways (SCAPs) [13][11]. SCAPs are redundant pro-survival pathways present in senescent cells, which downregulate key apoptotic modulators (e.g., caspases) [2,13][1][11]. This has the added benefit of broadening the therapeutic index, meaning the treatment has a broader efficacy with fewer side effects [13][11].
Metformin is a commonly used therapeutic for the treatment of diabetes mellitus and has been used since the 1950′s [65][45]. This drug has since garnered interest in the field of aging research given its association with decreased all-cause morbidity and mortality [66][46]. Metformin has senolytic and senotherapeutic activity through inhibition of the SASP and SCAPs [13][11]. In a 2010 study, metformin was used on the invertebrate Caenorhabditis elegans and promoted a longer lifespan with an increased health span [67,68][47][48]. This was also demonstrated in murine models [68][48]. Metformin is currently being investigated in the Targeting Aging with Metformin (TAME) clinical trial which aims to expand its indication to more broadly target age associated disease [74][54]. The TAME trial aims to compare patients being treated with metformin with patients who have stopped metformin treatments [74,75][54][55]. The occurrence of different age-related pathologies is then compared between each group of participants [74,75][54][55].
Senescent cells and the associated SASP can play different roles in different wound types. For example, certain senescent cell populations play a beneficial role in acute wound healing, so eliminating them could be detrimental to the healing of acute wounds. Further research is needed to investigate therapies that can be used to alleviate senescent cell load without disrupting the overall healing process.
References
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879.
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627.
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174.
- Wilkinson, H.N.; Hardman, M.J. Senescence in Wound Repair: Emerging Strategies to Target Chronic Healing Wounds. Front. Cell Dev. Biol. 2020, 8, 773.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The heterogeneity of cellular senescence: Insights at the single-cell level. Trends Cell Biol. 2022.
- Kirschner, K.; Rattanavirotkul, N.; Quince, M.F.; Chandra, T. Functional heterogeneity in senescence. Biochem. Soc. Trans. 2020, 48, 765–773.
- Wang, L.; Wang, B.; Gasek, N.S.; Zhou, Y.; Cohn, R.L.; Martin, D.E.; Zuo, W.; Flynn, W.F.; Guo, C.; Jellison, E.R.; et al. Targeting p21Cip1 highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2021, 34, 75–89.e8.
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310.
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beauséjour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.-Y.; Campisi, J. A Human-Like Senescence-Associated Secretory Phenotype Is Conserved in Mouse Cells Dependent on Physiological Oxygen. PLoS ONE 2010, 5, e9188.
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568.
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.-P.; Campeau, E.; Beauséjour, C.M.; Kim, S.-H.; et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81.
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.
- Kohli, J.; Wang, B.; Brandenburg, S.M.; Basisty, N.; Evangelou, K.; Varela-Eirin, M.; Campisi, J.; Schilling, B.; Gorgoulis, V.; Demaria, M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 2021, 16, 2471–2498.
- Pils, V.; Ring, N.; Valdivieso, K.; Lämmermann, I.; Gruber, F.; Schosserer, M.; Grillari, J.; Ogrodnik, M. Promises and challenges of senolytics in skin regeneration, pathology and ageing. Mech. Ageing Dev. 2021, 200, 111588.
- Wang, Z.; Shi, C. Cellular senescence is a promising target for chronic wounds: A comprehensive review. Burn. Trauma 2020, 8, tkaa021.
- Wang, B.; Liu, Z.; Chen, V.P.; Wang, L.; Inman, C.L.; Zhou, Y.; Guo, C.; Tchkonia, T.; Rowe, D.W.; Kuchel, G.A.; et al. Transplanting cells from old but not young donors causes physical dysfunction in older recipients. Aging Cell 2020, 19, e13106.
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189.
- Vasey, D.B.; Wolf, C.R.; Brown, K.; Whitelaw, C.B.A. Spatial p21 expression profile in the mid-term mouse embryo. Transgenic Res. 2010, 20, 23–28.
- Cho, A.-R.; Kim, J.Y.; Munkhbayer, S.; Shin, C.-Y.; Kwon, O. p21 upregulation in hair follicle stem cells is associated with telogen retention in aged mice. Exp. Dermatol. 2015, 25, 76–78.
- Safwan-Zaiter, H.; Wagner, N.; Wagner, K.-D. P16INK4A—More Than a Senescence Marker. Life 2022, 12, 1332.
- Behmoaras, J.; Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 2020, 220, e202010162.
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Doolittle, M.; Tchkonia, T.; Kirkland, J.L.; LeBrasseur, N.K.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. Targeted clearance of p21- but not p16- positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell 2022, 21, e13602.
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 11476.
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050.
- Alkema, W.; Boekhorst, J.; Eijlander, R.T.; Schnittger, S.; De Gruyter, F.; Lukovac, S.; Schilling, K.; Kortman, G.A.M. Charting host-microbe co-metabolism in skin aging and application to metagenomics data. PLoS ONE 2021, 16, e0258960.
- Chng, K.R.; Tay, A.S.L.; Li, C.; Ng, A.H.Q.; Wang, J.; Suri, B.K.; Matta, S.A.; McGovern, N.; Janela, B.; Wong, X.F.C.C.; et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat. Microbiol. 2016, 1, 16106.
- Oh, J.; Conlan, S.; Polley, E.C.; Segre, J.A.; Kong, H.H. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012, 4, 77.
- Dimitriu, P.A.; Iker, B.; Malik, K.; Leung, H.; Mohn, W.W.; Hillebrand, G.G. New Insights into the Intrinsic and Extrinsic Factors That Shape the Human Skin Microbiome. mBio 2019, 10, e00839-19.
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins Basic Pathology; Elsevier Health Sciences Division: Amsterdam, The Netherlands, 2017; Volume 10.
- Zeeuwen, P.L.; Boekhorst, J.; van den Bogaard, E.H.; de Koning, H.D.; van de Kerkhof, P.M.; Saulnier, D.M.; van Swam, I.I.; van Hijum, S.A.; Kleerebezem, M.; Schalkwijk, J.; et al. Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol. 2012, 13, R101.
- Potekaev, N.N.; Borzykh, O.B.; Medvedev, G.V.; Pushkin, D.V.; Petrova, M.M.; Petrov, A.V.; Dmitrenko, D.V.; Karpova, E.I.; Demina, O.M.; Shnayder, N.A. The Role of Extracellular Matrix in Skin Wound Healing. J. Clin. Med. 2021, 10, 5947.
- Jugé, R.; Rouaud-Tinguely, P.; Breugnot, J.; Servaes, K.; Grimaldi, C.; Roth, M.-P.; Coppin, H.; Closs, B. Shift in skin microbiota of Western European women across aging. J. Appl. Microbiol. 2018, 125, 907–916.
- Vukmanovic-Stejic, M.; Rustin, M.H.; Nikolich-Zugich, J.; Akbar, A.N. Immune responses in the skin in old age. Curr. Opin. Immunol. 2011, 23, 525–531.
- Kim, H.-J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748.
- Blair, M.J.; Jones, J.D.; Woessner, A.E.; Quinn, K.P. Skin Structure–Function Relationships and the Wound Healing Response to Intrinsic Aging. Adv. Wound Care 2020, 9, 127–143.
- Guo, S.; DiPietro, L.A. Factors Affecting Wound Healing. J. Dent. Res. 2010, 89, 219–229.
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419.
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell Biol. 2019, 97, 258–267.
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2020, 22, 75–95.
- Hardwick, J.M.; Soane, L. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722.
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47.
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in Skin Senescence Prevention and Treatment. Int. J. Mol. Sci. 2021, 22, 6814.
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576.
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44.
- Onken, B.; Driscoll, M. Metformin Induces a Dietary Restriction–Like State and the Oxidative Stress Response to Extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758.
- Soukas, A.A.; Hao, H.; Wu, L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol. Metab. 2019, 30, 745–755.
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256.
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83.
- Shi, J.; Zhou, Y.; Huang, H.-C.; Mitchison, T.J. Navitoclax (ABT-263) Accelerates Apoptosis during Drug-Induced Mitotic Arrest by Antagonizing Bcl-xL. Cancer Res. 2011, 71, 4518–4526.
- Wei, X.; Li, M.; Zheng, Z.; Ma, J.; Gao, Y.; Chen, L.; Peng, Y.; Yu, S.; Yang, L. Senescence in chronic wounds and potential targeted therapies. Burn. Trauma 2022, 10, tkab045.
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; DeMaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781.
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30.
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065.
More