The demand for new novel flavour and fragrance (F&F) molecules has boosted the need for a systematic approach to designing fragrance molecules. However, the F&F-related industry still relies heavily on experimental approaches or on existing databases without considering the consequences resulting from changes in concentration, which could omit potential fragrances. Computer-aided molecular design (CAMD) has great potential to identify novel molecular structures to be used as fragrances.
- fragrance molecules
- computer-aided molecular design
- rough sets
1. Introduction
2. Computer-Aided Molecular Design (CAMD)
3. Topological Indices (TIs)
4. Rough Set-Based Machine Learning (RSML)
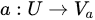
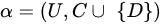
where C is the set of conditional attributes, and D is the decision attribute.
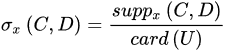
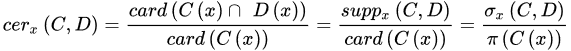
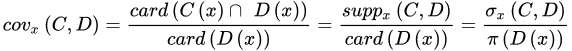
The strength represents the total number of samples that follow the generated rule divided by the total number of samples. The certainty factor is defined as the frequency of samples having the decision, D, in the sets of samples that fulfil conditions, C. Lastly, the coverage factor is the frequency of samples possessing conditions, C in the decision class. The former measures the predictive reliability of a rule, whilst the latter measures the generalisation power of a rule. A higher certainty indicates a lower chance of a molecule being misclassified, whereas a high coverage suggests that a rule is a good approximation of an underlying general principle. These three parameters will provide quantitative evidence to help select the most useful rule-based models.
References
- Fortune Business Insights. Flavors and Fragrances Market Size, Share Report (2021–2028). 2021. Available online: https://www.fortunebusinessinsights.com/flavors-and-fragrances-market-102329 (accessed on 3 April 2022).
- Sell, C.S. Chemistry and the Sense of Smell; John Wiley & Sons, Incorporated: Somerset, CA, USA, 2014.
- Zhang, L.; Mao, H.; Liu, L.; Du, J.; Gani, R. A machine learning based computer-aided molecular design/screening methodology for fragrance molecules. Comput. Chem. Eng. 2018, 115, 295–308.
- Korichi, M.; Gerbaud, V.; Floquet, P.; Meniai, A.H.; Nacef, S.; Joulia, X. Quantitative structure-Odor relationship: Using of multidimensional data analysis and neural network approaches. Comput. Aided Chem. Eng. 2006, 21, 895–900.
- Linke, P.; Kokossis, A. Simultaneous Synthesis and Design of Novel Chemicals and Chemical Process Flowsheets. In European Symposium on Computer Aided Process Engineering-12; Grievink, J., van Schijndel, Eds.; Elsevier: Amsterdam, The Netherlands, 2002; Volume 10, pp. 115–120.
- Austin, N.D.; Sahinidis, N.V.; Trahan, D.W. Computer-aided molecular design: An introduction and review of tools, applications, and solution techniques. Chem. Eng. Res. Des. 2016, 116, 2–26.
- Zhou, T.; Zhou, Y.; Sundmacher, K. A hybrid stochastic–deterministic optimization approach for integrated solvent and process design. Chem. Eng. Sci. 2017, 159, 207–216.
- Chemmangattuvalappil, N.G.; Ng, D.K.S.; Ng, L.Y.; Ooi, J.; Chong, J.W.; Eden, M.R. A review of process systems engineering (PSE) tools for the design of ionic liquids and integrated biorefineries. Processes 2020, 8, 1678.
- Liu, Q.; Zhang, L.; Tang, K.; Liu, L.; Du, J.; Meng, Q.; Gani, R. Machine learning-based atom contribution method for the prediction of surface charge density profiles and solvent design. AIChE J. 2021, 67, e17110.
- Zhang, L.; Mao, H.; Zhuang, Y.; Wang, L.; Liu, L.; Dong, Y.; Du, J.; Xie, W.; Yuan, Z. Odor prediction and aroma mixture design using machine learning model and molecular surface charge density profiles. Chem. Eng. Sci. 2021, 245, 116947.
- Mah, A.X.Y.; Chin, H.H.; Neoh, J.Q.; Aboagwa, O.A.; Thangalazhy-Gopakumar, S.; Chemmangattuvalappil, N.G. Design of bio-oil additives via computer-aided molecular design tools and phase stability analysis on final blends. Comput. Chem. Eng. 2019, 123, 257–271.
- Yee, Q.Y.; Hassim, M.H.; Chemmangattuvalappil, N.G.; Ten, J.Y.; Raslan, R. Optimization of quality, safety and health aspects in personal care product preservative design. Process Saf. Environ. Prot. 2022, 157, 246–253.
- Ooi, Y.J.; Aung, K.N.G.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Chemmangattuvalappil, N.G. Design of fragrance molecules using computer-aided molecular design with machine learning. Comput. Chem. Eng. 2022, 157, 107585.
- Chemmangattuvalappil, N.G. Development of solvent design methodologies using computer-aided molecular design tools. Curr. Opin. Chem. Eng. 2020, 27, 51–59.
- Zhang, L.; Mao, H.; Liu, Q.; Gani, R. Chemical product design—Recent advances and perspectives. Curr. Opin. Chem. Eng. 2020, 27, 22–34.
- Radhakrishnapany, K.T.; Wong, C.Y.; Tan, F.K.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Janairo, J.I.B.; Chemmangattuvalappil, N.G. Design of fragrant molecules through the incorporation of rough sets into computer-aided molecular design. Mol. Syst. Des. Eng. 2020, 5, 1391–1416.
- Brookes, J.C.; Horsfield, A.P.; Stoneham, A.M. Odour character differences for enantiomers correlate with molecular flexibility. J. R. Soc. Interface 2009, 6, 75–86.
- Islam, T.U.; Mufti, Z.S.; Ameen, A.; Aslam, M.N.; Tabraiz, A. On Certain Aspects of Topological Indices. J. Math. 2021, 2021, 9913529.
- Dearden, J.C. The use of topological indices in QSAR and QSPR modeling. Chall. Adv. Comput. Chem. Phys. 2017, 24, 57–88.
- Blay, V.; Gullón-Soleto, J.; Gálvez-Llompart, M.; Gálvez, J.; García-Domenech, R. Biodegradability Prediction of Fragrant Molecules by Molecular Topology. ACS Sustain. Chem. Eng. 2016, 4, 4224–4231.
- Amboni, R.D.D.C.; Junkes, B.D.; Yunes, R.A.; Heinzen, V.E.F. Quantitative structure—Odor relationships of aliphatic esters using topological indices. J. Agric. Food Chem. 2000, 48, 3517–3521.
- Chacko, R.; Jain, D.; Patwardhan, M.; Puri, A.; Karande, S.; Rai, B. Data based predictive models for odor perception. Sci. Rep. 2020, 10, 17136.
- Ham, C.L.; Jurs, P.C. Structure-activity studies of musk odorants using pattern recognition: Monocyclic nitrobenzenes. Chem. Senses 1985, 10, 491–505.
- Belyadi, H.; Haghighat, A. Introduction to machine learning and Python. In Machine Learning Guide for Oil and Gas Using Python; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–55.
- Dey, A. Machine Learning Algorithms: A Review. Int. J. Comput. Sci. Inf. Technol. 2016, 7, 1174–1179.
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215.
- Lundberg, S.M.; Lee, S.I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 4768–4777.
- Dobbelaere, M.R.; Plehiers, P.P.; van de Vijver, R.; Stevens, C.V.; van Geem, K.M. Machine Learning in Chemical Engineering: Strengths, Weaknesses, Opportunities, and Threats. Engineering 2021, 7, 1201–1211.
- Xu, G.; Papageorgiou, L.G. A mixed integer optimisation model for data classification. Comput. Ind. Eng. 2009, 56, 1205–1215.
- Zhang, Q.; Xie, Q.; Wang, G. A survey on rough set theory and its application. CAAI Trans. Intell. Technol. 2016, 1, 323–333.
- Pawlak, Z. Rough sets. Int. J. Comput. Inf. Sci. 1982, 11, 341–356.
- Pawlak, Z. Rough set approach to knowledge-based decision support. Eur. J. Oper. Res. 1997, 99, 48–57.
- Pawlak, Z. Some issues on rough sets. Lect. Notes Comput. Sci. 2004, 3100, 1–58.
- Mohamed, A.S.A. Application of rough set theory for clinical data analysis: A case study. Math. Comput. Model. 1991, 15, 19–37.
- Słowiński, K. Rough Classification of HSV Patients. Intell. Decis. Support. 1992, 11, 77–93.
- Tanaka, H.; Ishibuchi, H.; Matsuda, N. Fuzzy Expert System Based on Rough Sets and Its Application to Medical Diagnosis. Int. J. Gen. Syst. 1992, 21, 83–97.
- Aviso, K.B.; Janairo, J.I.B.; Promentilla, M.A.B.; Tan, R.R. Prediction of CO2 storage site integrity with rough set-based machine learning. Clean Technol. Environ. Policy 2019, 21, 1655–1664.
- Lei, L.; Chen, W.; Wu, B.; Chen, C.; Liu, W. A building energy consumption prediction model based on rough set theory and deep learning algorithms. Energy Build. 2021, 240, 110886.
- Raza, M.S.; Qamar, U. Rough Set Theory. In Understanding and Using Rough Set Based Feature Selection: Concepts, Techniques and Applications; Springer: Singapore, 2017; pp. 53–79.
- Pawlak, Z. Rough sets, decision algorithms and Bayes’ theorem. Eur. J. Oper. Res. 2002, 136, 181–189.