Cancer recurrence and metastasis, following successful treatment, constitutes a critical threat in clinical oncology and are the leading causes of death amongst cancer patients. This phenomenon is largely attributed to metastatic tumor dormancy, a rate-limiting stage during cancer progression, in which disseminated cancer cells remain in a viable, yet not proliferating state for a prolonged period. Dormant cancer cells are characterized by their entry into cell cycle arrest and survival in a quiescence state to adapt to their new microenvironment through the acquisition of mutations and epigenetic modifications, rendering them resistant to anti-cancer treatment and immune surveillance. Under favorable conditions, disseminated dormant tumor cells ‘re-awake’, resume their proliferation and thus colonize distant sites.
- metastasis
- dormancy
- tumor
1. Introduction
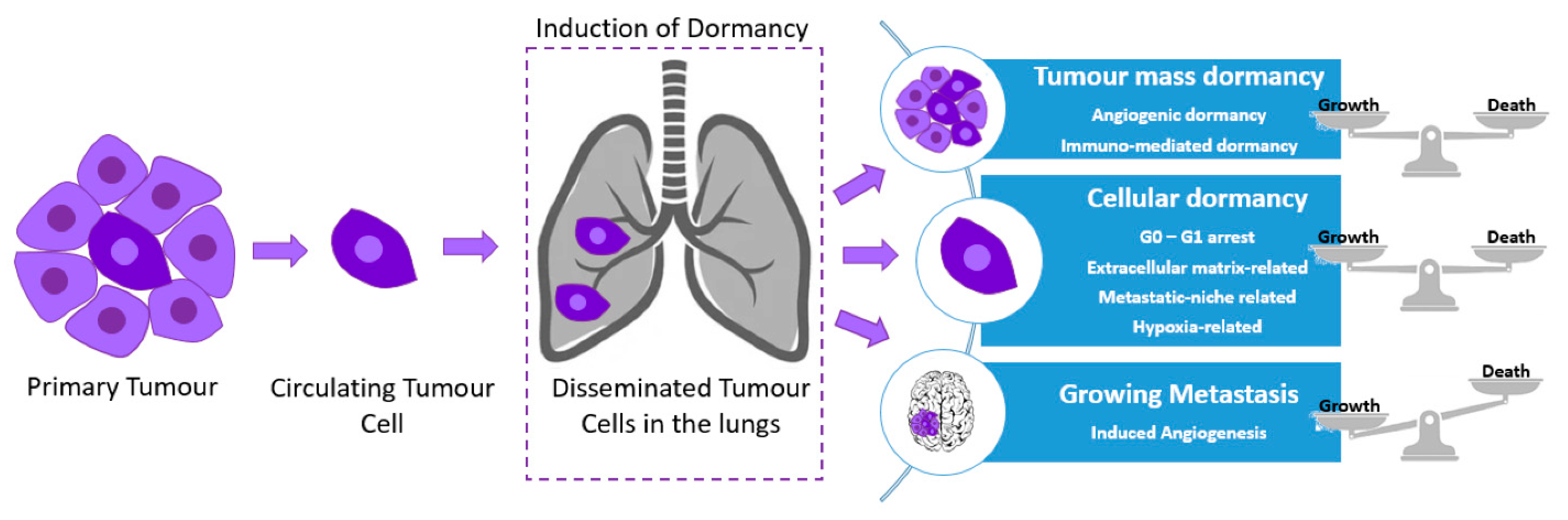
2. Mechanisms of Dormancy Induction and Maintenance
Dormancy and its maintenance are complex processes, mediated by intrinsic and autocrine signaling, and it is also controlled by signals derived from immune cells, endothelial cells, fibroblasts and other non-cellular components of the tumor microenvironment [23,24][16][17].
2.1. Intrinsic Mechanisms
2.1.1. Genetic Alterations
A correlation has been demonstrated between various alterations in the genome, primarily linked to cell differentiation/proliferation and the induction and maintenance of the dormant phenotype as shown in several preclinical cancer models. Overexpression of the FBXW7 gene has been rendered a crucial component for the maintenance of breast cancer dormancy. In the presence of FBXW7, entrance to the cell cycle is restrained through the ubiquitination and subsequent proteasomal degradation of its inducers c-Myc and cyclin E. When genetically ablated, the FBXW7 gene led to not only the disruption of the quiescent state of DTCs, but also the initiation of their proliferation in both allograft and xenograft models. DTCs lacking FBXW7 were significantly depleted following paclitaxel treatment, thus indicating increased sensitivity to chemotherapy. Poorer prognosis and shorter cell survival was associated with tumors expressing high levels of FBXW7 compared to FBXW7-deficient cells following tumor resection and chemotherapy treatment [25][18]. A dormant phenotype of breast DTCs in the bones has been linked to the leukemia inhibitory factor receptor (LIRF) which interacts with the interleukin-6 (IL-6) cytokine family. LIRF acts as a suppressor of cytokine signaling (SOCs) and an activator of signal transducer and activator transcription 3 (STAT3). Upon LIFR ablation, downregulation of dormancy-associated genes, reactivation of proliferation and bone colonization was observed [26][19]. Another gene associated with the maintenance of the dormant phenotype in head and neck squamous cell carcinoma (HNSCC) is the paired-related homeobox transcription factor (PRRX1). Its expression drives downregulation of mIR-642b-3p, which participates in tumorigenesis and cell growth and, therefore, sustains dormancy through p38 and transforming growth factor-β2 (TGFβ-2). Its implication with metastasis and invasion through the activation of the epithelial-to-mesenchymal transition program (EMT) indicates that this process could be exploited to sustain cancer cells in the dormant state [27][20]. Intraperitoneal and pulmonary metastasis is inhibited by kisspeptin 1 (KISS1), as shown in xenograft models of several cancer types, such as ovarian, breast and melanoma cancers [28][21]. The reverse role of protein kinase C (PKC) in re-activating cell migration, following inhibition by KISS1, is indicative of its potential as a molecular target for treatment [29][22]. In summary, genetic alterations play a crucial role in regulating the shift from quiescence to proliferation. Such genetic modifications have been found to not only reactivate cells but also to keep them in a suppressed steady state.2.1.2. Mechanisms of Mammalian Autophagy in Cancer Dormancy
Autophagy is another proposed crucial mechanism for the re-activation, survival and adaptation of dormant DTCs. Autophagy is an evolutionary conserved, physiological mechanism of fitness and cell survival, whose activation highly depends on the presence of unfavorable, metabolic stress conditions, such as nutrient deprivation. Its mechanism of action is associated with the degradation and subsequent recycling of damaged cytosol components, misfolded proteins, organelles and other macromolecules in order to sustain homeostasis and diminish cell damage. As such, evidence begins to accumulate regarding the exploitation of autophagy by tumor cells to endorse their survival under oxidative stress, improve their bioenergetics and thus facilitate tumor progression [30][23]. Proliferating cells generally show decreased levels of autophagy compared to dormant cells. Furthermore, an association has been made between the induction of a reversible dormancy state through activation of autophagy and the expression of the Aplasia Ras homolog member 1 (ARHI1) tumor suppressor gene in xenograft ovarian tumors. Tumor growth escalated following ARHI1 inhibition, suggesting a role in maintaining tumor dormancy. ARHI1 promotes autophagy through the upregulation of ATG4 cysteine protease and inhibition of the mammalian target of rapamycin and phosphatidylinositol 3-kinase-protein signaling pathway (PI3K/AKT/mTOR) [31][24]. It further enhances autophagy by driving nuclear localization of Forkhead box O3 (FOXO3a) and transcription factor EB (TFEB), which are pivotal mediators for the expression of various autophagy effectors, such as the microtubule-associated proteins 1A/1B light chain 3B (MAPLC3B) [22][25]. Contradicting results were acquired regarding the consequences of ARHI1 expression in vivo and in vitro, which led to cell dormancy and autophagic cell death, respectively. This is potentially attributed to survival signals induced by insulin growth factor 1 (IGF-1), interleukin 8 (IL-8) and vascular endothelial growth factor (VEGF) and their ability to release ovarian cancer cells from autophagic cell death in vitro. In contrast, these signals increased survival through delaying the outgrowth of the dormant ovarian tumor xenograft mouse models.2.1.3. Intracellular Signals
In the case of aggressive cancer development, tumor cells adapt to unique microenvironments by withstanding various cellular stresses. Various cell-derived factors have been proposed to act upon the modulation of signaling pathways between cell dormancy and cell growth. Consequently, intracellular signaling pathways are thought to affect the fate of cancer cells and drive the shift between their state of dormancy and tumorigenesis. The first signaling mechanism that has been associated with the dormant phenotype in many preclinical models involves the balance between p38α/β and the extracellular signal-regulated kinases (ERK1/2), part of the mitogen-activated protein kinase signaling pathway (MAPK) [34][26]. In fact, higher ratios were proposed to be analogous to tumor growth induction whereas lower ratios enhance tumor dormancy, with urokinase plasminogen activator receptor (uPAR) being a crucial modulator of this process [35][27]. The expression of uPAR in the presence of fibronectin activates both the epidermal growth factor receptor (EGFR) and alpha-5 beta-1 integrin [36][28]. Some of the mechanisms behind the ERK/p38 pathway, leading cells to a quiescent state, have been proposed following studies in a model of aggressive, human epidermoid-carcinoma cell line (HEp3). The following pathways correspond to cells exhibiting an ERK/p38 low ratio, a key characteristic of cell dormancy [37][29]. Cell cycle arrest in the G0-G1 phases and, therefore, entrance to the quiescent state can be regulated by overexpression of transcription factors p53, BHLHB3, NR2F1 and the downregulation of c-Jun and FOXM1 [34][26].
Cancer dormancy can be separated into cellular dormancy and tumor mass dormancy (rate of proliferation rate = apoptosis rate). Many intracellular factors can regulate tumor dormancy. An example of glioblastoma dormancy regulators include the epidermal growth factor receptor (EGFR) and thrombospondin (TSP) [38][30]. Other types of cancer exhibit recurrence years after treatment and surgical resection; however, depending on the neoplasm, the period of tumor latency significantly differs [39][31]. An increased likelihood of metastatic relapse at a distant site over a relatively short period of time, as well as increased levels of cell proliferation have been associated with triple-negative and human epidermal growth factor receptor 2 (HER2) breast cancers, when compared to other subtypes, indicative of their aggressive clinical course [40][32].
Another signaling pathway that has been investigated for its role in promoting tumor dormancy and the regulation of the metastatic phenotype in estrogen receptor-positive (ER+) breast cancer cells is NFkB. Following the expression of a ‘constitutively active form of IkB kinase b (CA-IKKβ)’ in cell lines, a reversible inhibition of E2-dependent cell proliferation and tumor growth was demonstrated in vitro and in vivo. Similarly, co-activation of IKKβ and ER enhanced cell invasion and migration to guide the experimental development of metastasis [46][33]. Decreased activity of the phosphatidylinositol 3′-kinase (PI3K)/AKT signaling pathway, comprising the extracellular stress protein clusterin and IGF1, under external stimuli is another mechanism thought to be involved in the switching between cancer cell proliferation and dormancy. The negative regulation of the pathway is primarily caused by the binding inhibition of IGF1 to its receptor and by the secretion of clusterin under serum deprivation conditions. Therefore, the cancer cell fate between dormancy and proliferation during tumor progression could be dictated by the interplay between IGF-1 and clusterin [47][34].
2.1.4. Epigenetic Mechanisms
Fluctuations between a proliferating and dormant state of DTCs could also be the consequence of epigenetic reprogramming mechanisms, encompassing transcriptional regulation through changes in chromatin structure. Some of those mechanisms include non-coding RNAs, whereas the majority of functions are exerted through histone modifications and DNA methylation [49][35]. In contrast to its hypermethylation-promoted downregulation observed in numerous cancers, overexpression of the orphan nuclear receptor (NR2F1) has been linked to dormancy of DTCs in prostate and HNSCC cancer patients. Cell quiescence activated by NR2F1 is controlled by the combination of cyclin-dependent kinase (CDK) inhibitors, retinoic acid receptor β (RARβ) and the SOX9 transcription factor. Chromatin repression is further enhanced by NR2F1 through the activation of NANOG, which is involved in the dormant state of DTCs within the bone marrow.
In ER+ breast cancer patients, the MSK1 kinase has been identified as another crucial regulator of metastatic dormancy, since its higher expression levels correspond to lower probability of developing early metastases. Breast cancer cell differentiation is impaired through the downregulation of MSK1, therefore enhancing their growth potential and bone metastases. The expression of the transcription factors FOXA1 and GATA3, which are vital for cell differentiation, is also regulated by MSK1 depending on the alterations of their chromatin status [52][36]. Conclusively, several mechanisms for epigenetic regulation of gene expression have been reported in dormant cancer cells. Each of these mechanisms hold unique characteristics that ensure reliable control of cell identity and phenotypic plasticity regarding the intrinsic and the extrinsic dormancy. Understanding the dynamic correlation between dormancy and the epigenome will improve ourthe knowledge for the molecular underpinnings of cell plasticity, and thus lead to the identification of new novel targets for the treatment of metastatic cancers.2.2. Tumor Microenvironment
Tumors comprise the ECM and a complex mass of cellular components (non-malignant and malignant cells) which are conjointly referred to as tumor microenvironment (TME). Ectopic, uncontrolled growth is hindered by mechanisms encoded in all adult tissues, therefore, regulatory mechanisms that contribute to dormancy onset and the prevention of DTCs expansion are assumed to be encoded by tumor-naive target organ microenvironments. Initiation and maintenance of tumor dormancy is, hence, highly dependent on the crosstalk between cancer cells and the TME [53][37].2.2.1. Extracellular Matrix (ECM)
The interplay between cancer cells and their microenvironment strongly influences tumor progression with respect to tumor metastasis, annihilation or induction of dormancy [54][38]. P38/ERK ratio plays a catalytic role during switching between the dormant and proliferative state through the interaction with the ECM, which is a crucial TME component[55][39]. The simultaneous α5β1 integrin and urokinase-plasminogen activator receptor (uPAR)-driven ERK activation and the fibronectin-mediated p38 suppression spawn the proliferative nature of human epidermoid carcinoma (HEp3) cells [56][40]. Growth suppression and therefore the induction of a dormant cell phenotype, however, can be induced by any imbalance between this interaction. P38 hindrance alters this balance followed by a subsequent release of dormant cells into a proliferative state. Hypoxia and ER stress could also compel a pro-survival mechanism following weakened folding of proteins [57][41]. B1-integrin suppression has also been linked to the induction of dormancy, as shown in in vitro studies, and using in vivo mouse models for breast cancer. The presence of actin-stress fibers and fibronectin is triggered through integrin activation, thus further elucidating the significance of the TME and ECM in promoting cellular dormancy [58][42].
2.2.2. Hypoxia and Angiogenesis
Tumor survival depends on angiogenesis, since tumors unable to intravasate are driven either to apoptosis or in a quiescent, dormant state until the acquisition of sufficient additional alterations, allowing them to escape and restart proliferating [60][43]. An equivalent rate between programmed cell death and cell proliferation determines metastases and the maintenance of tumor dormancy. The indirect increase in apoptotic rate in tumor cells through inhibitors of angiogenesis regulates metastatic growth [20][44]. A hypoxic TME, a key feature of many solid tumors, is a result of oxygen depletion and therefore a disrupted homeostasis [62][45]. Hypoxia has been proposed as a significant driver of tumor dormancy, eliciting a DTC subpopulation to enter the dormant state and enable adaptation and survival for a long period of time [63][46]. Endothelial cells (ECs) produce signals that regulate dormancy in cancer cells, with DII4 (Notch ligand) being a prime example. In ECs, DII4 has been associated with the escape of human colorectal or T-cell acute lymphoblastic leukemia (T-ALL) cells from dormancy, consistent with its role in tumor angiogenesis and the positive regulation of the Notch signaling pathway. In contrast to its absence within quiescent tumors, increased expression of DII4 in aggressive tumors provides further evidence for its contribution in tumor dormancy [65][47]. Activation of Notch 3 by angiogenic factors in ECs, stimulates a tumorigenic phenotype. While Notch 3 consistently displays decreased expression in dormant tumors, phosphorylated p38, a target of the canonical phosphatase MKP-1 controlled by Notch, was found to be present in increased levels within dormant cells. Therefore, tumor dormancy can be regulated by angiogenesis-driven mechanisms, which include the MAPK and Notch pathways [66][48].2.2.3. Immune System
The immune system is another fundamental regulator for the induction and maintenance of tumor dormancy [24][17]. Alterations accumulating in tumor cells can make them genetically unstable and render them resistant to complete recognition and elimination by the immune system components [76][49]. The immune system has been indirectly linked to tumor dormancy following observations of increased immune cell infiltration in breast cancer patients with DTCs in the bone marrow. T cell content and immune cell activation in tumors in the bone marrow of cancer patients was significantly increased compared to healthy individuals, indicating signs of tumor dormancy. In other words, the bone marrow represented a prosperous site for the presence of lethal DTCs in a dormant state with a potential contribution to this by the high presence of T cells [76][49]. The maintenance of dormancy in a murine B cell lymphoma model (BCL1) required the presence of cytostatic CD8+ T-cells, the depletion of which reversed cancer cell dormancy [77][50]. Similarly, in a mouse melanoma model, cancer cell dormancy in the lung was found to be governed by CD8+ T cells, the decrease in which caused visceral metastases and faster outgrowth, providing further evidence that metastatic growth is regulated by the immune system [78][51]. The dormant state of metastatic cells can also be maintained by T lymphocytes, through interferon-γ (IFN-γ) production that was shown to be able to arrest tumor cells to the G0/G1 phase [79][52]. Cellular dormancy could also be influenced by immune evasion. In a model of acute myeloid leukemia, overexpression of the programmed death-ligand 1 (PD-L1) (also known as B7-H1) led to cytotoxic T cell-mediated resistance to killing and thus a potential contribution dormancy [80][53]. Dormancy could be also potentiated following the entrance of DTCs in immune-privileged stem cell niches where high levels of regulatory T cells could accumulate under specific circumstances (Tregs) as described by [81][54]. The role of the immune system in controlling tumor cell dormancy in primary and metastatic foci has been underlined by several studies. The current discovery of new immune checkpoint inhibitors has marked a new era of hope for cancer therapy. Nevertheless, the development of immunotherapy against dormant cells is puzzling, as quiescent DTCs have developed new unknown mechanisms to evade the immune system. More therapies that mobilize the immune system against dormant DTCs need to be developed. Despite the current studies, there is still a lot of room to clarify the mechanisms involved in immune-mediated induction of dormancy in tumor cells. This includes understanding how tumor evasion and dormancy are coupled or not.3. Escape from Dormancy
Cancer cells originating from a primary tumor mass persist in a clinically undetectable, dormant state for a protracted period of time, following their dissemination to distant sites. This is a consequence of the new, often unfavorable microenvironment they encounter during their residency at distant regions, causing their entry into senescence or dormant state. Dormancy entry is mediated by either a withdrawal from the proliferative cell cycle or the achievement of equilibrium between apoptosis and proliferation [30,95][23][55]. In the meantime, the accumulation of genetic and epigenetic aberrations renders DTCs fully adapted to the new microenvironment. A subgroup of DTCs escape their dormant state and ‘re-awake’, moving on to the formation of expanding masses, in response to not yet fully characterized cues [7]. Dormancy escape could be stimulated through modifications in the TME which comprises a heterogeneous population of non-cancer (e.g., pericytes, cancer-associated fibroblasts, tumor-associated macrophages, lymphocytes and leukocytes), cancer cells and the ECM, encompassed by the secretion of numerous molecules, including metabolites, growth factors, cytokines and lymphokines. Through the constantly evolving interaction with TME components, the fate and behavior of cancer cells are dictated depending on the balance between proliferation and quiescence [63][46]. At the metastatic site, dormant breast cancer cells shift to proliferation following the displacement of type-1 collagen via β1-integrin signaling pathways, also affecting cytoskeletal architecture [96][56]. The brain, bone marrow and lung microvasculature consist of residing dormant cells. Quiescence is favored by the secretion of the anti-angiogenic factor thrombospondin-1 (TSP-1), however, during neovascularization, this suppressive signal is lost and TSP-1 switches to promoting cancer cell proliferation and significantly accelerating tumor growth rates. Therefore, a dormant niche is associated with a stable microvasculature, whereas outgrowing metastases are linked to neovasculature formation. Furthermore, several tumor-promoting factors secreted from the endothelial cells, such as periostin and TGF-β1, were identified as major inducers of new blood vessel formation in head and neck cancer [97][57]. Moreover, lipid mediators, such as the fatty-acid transporter CD36, can bind collagen or TSP-1 and actively control escape from cancer dormancy, as previously shown in metastatic prostate and oral cancer cells [98,99][58][59]. CD36-mediated tumor growth is achieved through lipid oxidation stimulated by the uptake of fatty acids [100][60]. Along with cytoskeletal re-organization, various elements of the ECM dictate dormancy escape. Another proposed mechanism involved in tumor escape from dormancy is chronic inflammation of the host tissue. An example is chronic inflammation generated by either smoking cigarettes or from liposaccharides derived from bacteria, both of which drive neutrophil activation and therefore the formation of ‘neutrophil extracellular traps’ (NETs). NETs are crucial for the re-activation of DTC proliferation, as they secrete matrix metalloproteinase 9 (MMP-9) and neutrophil elastase (NE) following neutrophil degradation. Protease secretion amplifies ‘basement membrane laminin-111’ expression, which interacts with the α3β1 integrin [106][61]. Re-awakening and re-activation of breast DTCs proliferation could also be attributed to monocyte chemoattractant protein-1 (MCP-1), interleukin-8 (IL-8) or the secretion of other soluble factors from the hepatic stellate cells (HSCs), located in the liver, induced via the ERK pathway [108][62]. When it comes to the bone marrow microenvironment, DTCs exited their dormant state following the overexpression of inflammatory cytokines or upon other alterations within the bone secretome, such as interactions with specialized tissue-specific cells and nutrients/metabolites [109][63]. Mechanisms that mediate dormant cell re-awakening might differ depending on the metastatic niche they occupy as each microenvironment responds to different challenges. DTCs exist in their dormant state for a protracted period of time in areas that are highly vascularized, such as the bone marrow [110][64]. Their escape is highly imposed by the activation of osteoclasts [111][65]. Following osteoclast recruitment and activation by VCAM-1, a chain of events takes place, including tumor expansion and bone destruction [112][66]. Activation of TGFβ1 or interleukin-8 (IL-8) and interleukin-6 (IL-6) secretion could be activated upon stromal injuries induced in the bone marrow and therefore reactivate dormant breast cancer cells [113][67]. Myeloma dormant cells were greatly reduced following resorption of osteoclasts by the receptor-activator nuclear factor-kb ligand (RANKL) [114][68]. Similarly, the formation of bone metastases initiated from dormant prostate and breast cancer cells was evident upon castration/ovariectomy [115][69]. Various proliferation-promoting mechanisms have been implicated in the lung with regards to the escape of dormant breast cancer cells. Some of these include initiation of proliferation by the activation of tank-binding kinase-1 (TBK1) [116][70], as well as the overexpression of periostin, driving Wnt signaling. Depending on the target organ, distinct mechanisms of escape from dormancy are implicated. This is evident by the effects of Coco, a TGFβ1 ligand antagonist, in metastatic sites of the lung, resulting in re-activation of breast cancer cells, but not in brain or bone metastases [117,118][71][72].4. Therapeutic Implications of Tumor Dormancy/Treatment Strategies
The likelihood of relapse could be significantly decreased upon application of therapeutic interventions directly targeting tumor dormancy. During cancer progression, dormant tumor cells often co-exist with rapidly proliferating cells, thus contributing to the development of therapeutic resistance [120,121,122][73][74][75]. Conventional therapies almost exclusively target fast-proliferating cells, rendering dormant cells resistant to those treatments. The eradication of tumor cells could be feasible through the identification of novel therapeutic approaches by unraveling the mechanisms responsible for controlling the transformation between proliferation and dormancy [23][16]. Mechanisms focusing on differentiating dormant cells from other non-proliferative yet viable cells, through the identification of their distinct characteristics, could be promising. However, despite the evident contribution of dormant cells in promoting metastatic disease relapse, their targeting remains unsuccessful [123][76].4.1. Maintaining Tumor Dormancy
A core therapeutic strategy that has been proposed aims to permanently sustain the dormant state of cells, maintaining residual cells non-proliferative and inactive. Hormone therapy is a promising cytostatic approach which drives cells into quiescence and proliferation arrest through entrance in a cell cycle G0-G1 phase [123][76]. Similarly, the use of palbociclib, abemaciclib, ribociclib or other CDK4/6 cell cycle inhibitors, which play crucial roles in G1/S phase transition, could promote cancer cell dormancy maintenance [124][77]. Another approach tested in HEp3 cells involves the inhibition of urokinase-type plasminogen activator receptor (uPAR) signaling. Tumorigenesis is induced in the presence of uPAR since it activates the α5β1 integrin that has a pivotal role in ERK activation by initiating an intracellular signaling cascade via Src and focal adhesion kinase (FAK) [125][78]. The activities of Src kinase and ERK, both activated when uPAR is overexpressed, can also be inhibited to prevent the formation of metastatic lesions or avoid dormant cell proliferating outbreaks by promoting translocation of cyclin-dependent kinase inhibitor p27 in the nucleus [126][79]. Dormant breast DTCs were maintained with both PP1 (Src inhibitor) and U0126 (ERK inhibitor) [38,127][30][80].
4.2. Re-Awakening and Sensitization of Dormant Cells to Therapy
Another therapeutic approach focuses on increasing susceptibility of dormant cells to anti-cancer treatment by disturbing their dormant state and ‘re-awakening’ them. Dormant cell re-entrance into the G2-M phase of the cell cycle would therefore lead to increased sensitivity towards cytotoxic therapy [130][81]. Reactivation of the cell cycle in dormant cells can be achieved by targeting microenvironment components that promote a dormant phenotype. Osteopontin neutralization, which is secreted in the bone marrow by osteoblasts, drove dormant leukemia cells to enter the cell cycle and initiate proliferation. Residual disease was considerably decreased and killing of neoplastic cells was achieved through the synergistic effect of osteopontin inhibition with cytarabine, a chemotherapeutic anti-metabolite [131][82]. Re-entrance of quiescent leukemia cells into the cell cycle was made possible through the act of granulocyte colony stimulating factor (G-CSF) which enhanced cytarabine cytotoxicity [132][83].4.3. Direct Targeting of Dormant Cells
Another therapeutic approach aims at directly targeting dormant cells to reduce the possibility of future disease relapse [10]. A worse patient survival rate has been previously associated with the suppression of autophagy pathways strictly in dormant breast cancer cells [137][84]. However, metastatic outgrowth was only minimally affected by the suppression of autophagy in cells that had already switched to a proliferating state. This indicates that administration of autophagy inhibitors is more effective during the tumor latency phase rather than during the cell proliferative phase. Treatment of dormant cells using epigenetic therapies is also being explored through the suppression of epigenetic enzymes, such as histone deacetylases (HDACs) and lysine demethylases (KDMs) [138][85]. Persistent, drug-resistant cancer cell survival requires histone 3 (H3) demethylation on lysine 4 (H3K4), catalyzed by a family of histone demethylases referred to as KDM5. A variety of KDM inhibitors currently exist, among which GSK-J4 is the most commonly used, targeting KDM2B [139][86], KDM5 [140][87] and KDM6A/B [141][88]. Dormant lung and glioblastoma DTCs were eliminated by GSK-J4, following taxane–platinum-based chemotherapy and dasatinib treatment, respectively [142,143][89][90]. Another inhibitor exhibiting increased specificity for KDM5 is CPI-455, whose mechanism of action includes an increase in H3K4 trimethylation (H3K4me3) levels and reduced Drug-Tolerant Persister cancer cell (DTPs) numbers in various cancer cell lines that have undergone chemotherapy treatment. Successful DTC killing by CPI-455 was shown in breast, colon, melanoma and lung cancer models [144][91].4.4. Advances in Targeting DTCs
Therapeutic targeting of dormant DTCs has therefore attracted significant interest and constitutes a promising avenue for improved clinical outcomes of cancer patients. Unfortunately, due to the immense amount of information missing regarding cellular or surface markers that could lead to the identification of DTCs as well as limitations in current diagnostic technologies, this therapeutic strategy is still very challenging [147][92]. A permanent state of dormancy and therefore blocking of metastases might be achieved by interfering with the balance between ERK and p38 status, the alteration of which links to various networks and dormancy escape-promoting signaling cascades [36][28]. The balance of p38/ERK could be altered through the functions of integrins and fibronectin, the binding of which is mediated by periostin, tenascin, macrophages or other ECM components, such as cytokines and growth factors, leading to DTCs re-awakening. Proteinase enzymes, secreted from neutrophils, lead to ECM remodeling and signaling integration, leading to DTC re-awakening [147][92].4.5. Future Therapeutic Interventions
The acceptance and understanding of the mechanisms that drive colony formation during cancer metastasis is fundamental. New mechanistic studies can be designed in order to look forward and produce new advanced models appropriate to study the development of metastasis from early, genomic immature DCCs [158][93]. Naume and colleagues managed to identify DCCs as a surrogate marker for adjuvant treatment effects. Such studies must be linked to other therapeutic attempts, such as omics analysis of DCCs and the evaluation of the pre-therapeutic and post-therapeutic microenvironment [159][94]. Through such combinations, scholars will gain extensive insights for the development of drugs that prevent activation of dormant DCCs, target slow-growing DCCs, evolving or stimulated DCCs and explosively growing DCCs. Thus, the role of the microenvironment in metastatic colony formation and growth is of paramount importance and should inspire the search for and clinical testing of drugs that either kill DCCs without generating inflammation, senescence and without withdrawing vital stimuli provided by the microenvironment [158][93]. The implementation of such combinatorial methods remains a major challenge, which can become even greater when it comes to testing these novel metastasis prevention models in the clinic. The available methods to detect DCCs are currently not sensitive enough to use eradication of DCCs as a readout, making it very difficult and time consuming to perform follow-up studies. Consistent discovery of early systemic cancer, molecular risk assessment and identification of predictive markers during the invisible phase of metastatic progression should become an intensified research field in the near future. Using good substitute end-points, the implementation of adjuvant therapy studies, delivered from trial designs used for patients with evident metastasis, may become more feasible and pave the way for balanced advances of adjuvant therapies [158][93]. Overall, dormant tumor cells are characterized as promising targets for the prevention of metastasis. The classification of different growth-arrested cell populations is not clear, and future research is necessary to separate their phenotypes. Conventional therapies do not target dormant tumor cells, thus there is an urgent need to combine the available conventional therapies or study if higher dosages of these available treatments may eliminate dormant tumor cells [19][95]. A spin in drug penetration research, a major challenge in cancer treatment, is necessary to give escalation to next-generation therapies. Modern nanotechnological methodologies may accomplish this necessity. The in vivo validation of novel therapies is important to approve feasibility of dormant tumor cell eradication by nanomedicine strategies. As tumor masses consist of highly heterogeneous cell populations, conventional therapies can possibly be supplemented with innovative dormant tumor cell-targeted probes [19][95].References
- Conti, C.J. 14.16—Mechanisms of Tumor Progression. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2010; pp. 335–347.
- Fidler, I.J. The biology of cancer metastasis. Semin. Cancer Biol. 2011, 21, 71.
- Friberg, S.; Nyström, A. Cancer Metastases: Early Dissemination and Late Recurrences. Cancer Growth Metastasis 2015, 8, 43–49.
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846.
- Goss, P.E.; Chambers, A.F. Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer 2010, 10, 871–877.
- Hadfield, G. The dormant cancer cell. Br. Med. J. 1954, 2, 607.
- Kareva, I. Escape from tumor dormancy and time to angiogenic switch as mitigated by tumor-induced stimulation of stroma. J. Theor. Biol. 2016, 395, 11–22.
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E. CD4+ T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498.
- Schardt, J.A.; Meyer, M.; Hartmann, C.H.; Schubert, F.; Schmidt-Kittler, O.; Fuhrmann, C.; Polzer, B.; Petronio, M.; Eils, R.; Klein, C.A. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell 2005, 8, 227–239.
- Osisami, M.; Keller, E.T. Mechanisms of metastatic tumor dormancy. J. Clin. Med. 2013, 2, 136–150.
- Furuya, M.; Yonemitsu, Y. Cancer neovascularization and proinflammatory microenvironments. Curr. Cancer Drug Targets 2008, 8, 253–265.
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132.
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer 2008, 8, 329–340.
- Badia-Ramentol, J.; Linares, J.; Gómez-Llonin, A.; Calon, A. Minimal residual disease, metastasis and immunity. Biomolecules 2021, 11, 130.
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572.
- Neophytou, C.M.; Kyriakou, T.C.; Papageorgis, P. Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 6158.
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622.
- Shimizu, H.; Takeishi, S.; Nakatsumi, H.; Nakayama, K.I. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight 2019, 4, 21.
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089.
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.S.; Wu, J.S.; Yu, X.H.; Wu, J.B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia 2019, 21, 216–229.
- Lee, J.H.; Welch, D.R. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997, 57, 2384–2387.
- Jiang, Y.; Berk, M.; Singh, L.S.; Tan, H.; Yin, L.; Powell, C.T.; Xu, Y. KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin. Exp. Metastasis 2005, 22, 369–376.
- Vera-Ramirez, L.; Hunter, K.W. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Res 2017, 6, 2134.
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929.
- Sutton, M.N.; Yang, H.; Huang, G.Y.; Fu, C.; Pontikos, M.; Wang, Y.; Mao, W.; Pang, L.; Yang, M.; Liu, J.; et al. RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 2018, 14, 637–653.
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672.
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879.
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695.
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857.
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250.
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; Van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279.
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434.
- El-Shennawy, L.; Dubrovskyi, O.; Kastrati, I.; Danes, J.M.; Zhang, Y.; Whiteley, H.E.; Creighton, C.J.; Frasor, J. Coactivation of Estrogen Receptor and IKKβ Induces a Dormant Metastatic Phenotype in ER-Positive Breast Cancer. Cancer Res. 2018, 78, 974–984.
- Jo, H.; Jia, Y.; Subramanian, K.K.; Hattori, H.; Luo, H.R. Cancer cell-derived clusterin modulates the phosphatidylinositol 3’-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol. Cell Biol. 2008, 28, 4285–4299.
- Robinson, N.J.; Parker, K.A.; Schiemann, W.P. Epigenetic plasticity in metastatic dormancy: Mechanisms and therapeutic implications. Ann. Transl. Med. 2020, 8, 177.
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 2018, 20, 211–221.
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71.
- Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305.
- Bichsel, C.A.; Wang, L.; Froment, L.; Berezowska, S.; Müller, S.; Dorn, P.; Marti, T.M.; Peng, R.W.; Geiser, T.; Schmid, R.A.; et al. Increased PD-L1 expression and IL-6 secretion characterize human lung tumor-derived perivascular-like cells that promote vascular leakage in a perfusable microvasculature model. Sci. Rep. 2017, 7, 10636.
- Parker, A.L.; Cox, T.R. The Role of the ECM in Lung Cancer Dormancy and Outgrowth. Front. Oncol. 2020, 10, 1766.
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157.
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100.
- Pisco, A.O.; Brock, A.; Zhou, J.; Moor, A.; Mojtahedi, M.; Jackson, D.; Huang, S. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat. Commun. 2013, 4, 2467.
- McNichols, D.W.; Segura, J.W.; DeWeerd, J.H. Renal cell carcinoma: Long-term survival and late recurrence. J. Urol. 1981, 126, 17–23.
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844.
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323.
- Indraccolo, S. Insights into the regulation of tumor dormancy by angiogenesis in experimental tumors. Adv. Exp. Med. Biol. 2013, 734, 37–52.
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25.
- Farrar, J.D.; Katz, K.H.; Windsor, J.; Thrush, G.; Scheuermann, R.H.; Uhr, J.W.; Street, N.E. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. J. Immunol. 1999, 162, 2842–2849.
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039.
- Wang, H.F.; Wang, S.S.; Huang, M.C.; Liang, X.H.; Tang, Y.J.; Tang, Y.L. Targeting Immune-Mediated Dormancy: A Promising Treatment of Cancer. Front. Oncol. 2019, 9, 498.
- Saudemont, A.; Quesnel, B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 2004, 104, 2124–2133.
- Zou, L.; Barnett, B.; Safah, H.; Larussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004, 64, 8451–8455.
- Willis, R.A. The Spread of Tumors in the Human Body; Butterworth: London, UK, 1952.
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456.
- Qin, X.; Yan, M.; Zhang, J.; Wang, X.; Shen, Z.; Lv, Z.; Li, Z.; Wei, W.; Chen, W. TGFβ3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci. Rep. 2016, 6, 20587.
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45.
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758.
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 017–0646.
- Hoskin, P.J. Hypoxia dose painting in prostate and cervix cancer. Acta Oncol. 2015, 54, 1259–1262.
- Johnson, R.W.; Schipani, E.; Giaccia, A.J. HIF targets in bone remodeling and metastatic disease. Pharmacol. Ther. 2015, 150, 169–177.
- Qiu, B.; Simon, M.C. Oncogenes strike a balance between cellular growth and homeostasis. Semin. Cell Dev. Biol. 2015, 43, 3–10.
- Eckers, J.C.; Kalen, A.L.; Sarsour, E.H.; Tompkins, V.S.; Janz, S.; Son, J.M.; Doskey, C.M.; Buettner, G.R.; Goswami, P.C. Forkhead box M1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat. Res. 2014, 182, 420–429.
- Bragado, P.; Sosa, M.S.; Keely, P.; Condeelis, J.; Aguirre-Ghiso, J.A. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012, 195, 25–39.
- Weidenfeld, K.; Schif-Zuck, S.; Abu-Tayeh, H.; Kang, K.; Kessler, O.; Weissmann, M.; Neufeld, G.; Barkan, D. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget 2016, 7, 71362–71377.
- Gammon, L.; Biddle, A.; Heywood, H.K.; Johannessen, A.C.; Mackenzie, I.C. Sub-sets of cancer stem cells differ intrinsically in their patterns of oxygen metabolism. PLoS ONE 2013, 8, e62493.
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983.
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932.
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903.
- Gammon, L.; Mackenzie, I.C. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J. Oral. Pathol. Med. 2016, 45, 77–82.
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779.
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337.
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arqués, O.; Martínez-Quintanilla, J.; Cuesta-Borrás, E.; Ramírez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905.
- Wolter, K.; Zender, L. Therapy-induced senescence—An induced synthetic lethality in liver cancer? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 135–136.
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141.
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430.
- Aguirre Ghiso, J.A. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene 2002, 21, 2513–2524.
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168.
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716.
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2+ cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47.
- Achen, M.G.; Stacker, S.A. Molecular control of lymphatic metastasis. Ann. N. Y. Acad. Sci. 2008, 1131, 225–234.
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 2010, 28, 275–280.
- Mascré, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Broheé, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262.
- Arosio, A.D.; Pignataro, L.; Gaini, R.M.; Garavello, W. Neck lymph node metastases from unknown primary. Cancer Treat. Rev. 2017, 53, 1–9.
- Staberg, M.; Rasmussen, R.D.; Michaelsen, S.R.; Pedersen, H.; Jensen, K.E.; Villingshøj, M.; Skjoth-Rasmussen, J.; Brennum, J.; Vitting-Seerup, K.; Poulsen, H.S.; et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 2018, 12, 406–420.
- Heinemann, B.; Nielsen, J.M.; Hudlebusch, H.R.; Lees, M.J.; Larsen, D.V.; Boesen, T.; Labelle, M.; Gerlach, L.O.; Birk, P.; Helin, K. Inhibition of demethylases by GSK-J1/J4. Nature 2014, 514, E1–E2.
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408.
- Dalvi, M.P.; Wang, L.; Zhong, R.; Kollipara, R.K.; Park, H.; Bayo, J.; Yenerall, P.; Zhou, Y.; Timmons, B.C.; Rodriguez-Canales, J.; et al. Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors. Cell Rep. 2017, 19, 1669–1684.
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538.
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581.
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694.
- Naume, B.; Synnestvedt, M.; Falk, R.S.; Wiedswang, G.; Weyde, K.; Risberg, T.; Kersten, C.; Mjaaland, I.; Vindi, L.; Sommer, H.H.; et al. Clinical outcome with correlation to disseminated tumor cell (DTC) status after DTC-guided secondary adjuvant treatment with docetaxel in early breast cancer. J. Clin. Oncol. 2014, 32, 3848–3857.
- Damen, M.P.F.; van Rheenen, J.; Scheele, C. Targeting dormant tumor cells to prevent cancer recurrence. FEBS J. 2021, 288, 6286–6303.