Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Muhammad Shahid Javaid.
The challenges in making animal models of complex human epilepsy phenotypes with varied aetiology highlights the need to develop alternative disease models that can address the limitations of animal models by effectively recapitulating human pathophysiology. The advances in stem cell technology provide an opportunity to use human iPSCs to make disease-in-a-dish models.
- stem cell lines
- epilepsy
- patient-specific cell models
- neuronal differentiation
- personalised disease models
- personalised medicine
1. Introduction
Epilepsy affects 70 million people worldwide [1[1][2],2], causing significant morbidity and mortality. with more than half of the affected people living in countries with poor medical resources and little or no access to treatment [3]. It is characterised by unprovoked, recurrent seizures which result from the disruption in the balance between neuronal excitation and inhibition in the brain [1]. In most cases the cause of epilepsy is unknown but both genetic and environmental factors are understood to contribute to its aetiology.
Genetic epilepsy is characterised by seizures that are the result of known genetic variance in one or many genes associated with epilepsy [2]. Mutations in several genes encoding ion channels and proteins have been reported to be most commonly associated with epilepsy. These genes include, but are not limited to mutation in SCN1A (encode sodium channel protein), which is associated with Dravet syndrome; KCNQ2 or KCNQ3 (both encoding potassium channel protein), which are associated with benign neuronal familial seizures [3,4,5][3][4][5]; the CHRNA4 gene (20q13), which is associated with Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE), characterised by hypermotor nocturnal seizures [6]; and the mammalian target of rapamycin (mTOR) pathway, in particular DEPDC5 gene of this pathway, which are associated with Focal Cortical Dysplasia (FCD) type IIa and IIb [7,8,9,10,11,12,13,14,15][7][8][9][10][11][12][13][14][15].
Anti-seizure medications (ASMs) are the mainstay of treatments for epilepsy. Despite multiple newly developed drug treatments being introduced to clinical practice, more than 30% of epileptic patients remain drug-resistant [16]. Genetic causes account for almost 20% of drug-resistant epilepsy cases in children. Surgery may be the only curative treatment option for these refractory epilepsy patients. However, ASMs and surgery are not always successful due to an incomplete understanding of epilepsy aetiology and pathogenesis resulting in non-targeted treatments [16,17,18,19][16][17][18][19]. There is, therefore, a need for in-depth investigations to gain a better understanding of the pathological mechanisms, an understanding which would inform the development of new treatments.
Currently, potential ASMs are validated using acute-seizure animal models prior to clinical development. However, animal models of genetic epilepsy have several limitations [20,21,22,23][20][21][22][23]. The major concern is the species-specific differences [23] leading to differences in physiological development and lack of human-specific receptors and drug targets. Hence, there is a demanding need for human-based disease models to develop new therapeutic strategies to achieve seizure freedom for these patients.
The development of human-based in vitro disease models [24] is an active area of research. One potential source of human in vitro models is the use of human pluripotent stem cells. These models can either be derived from human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs). The iPSCs derived from healthy individuals or patients are a promising approach to developing regenerative therapies as well as in vitro models of pathophysiological mechanisms of diseases (Figure 1) [25].
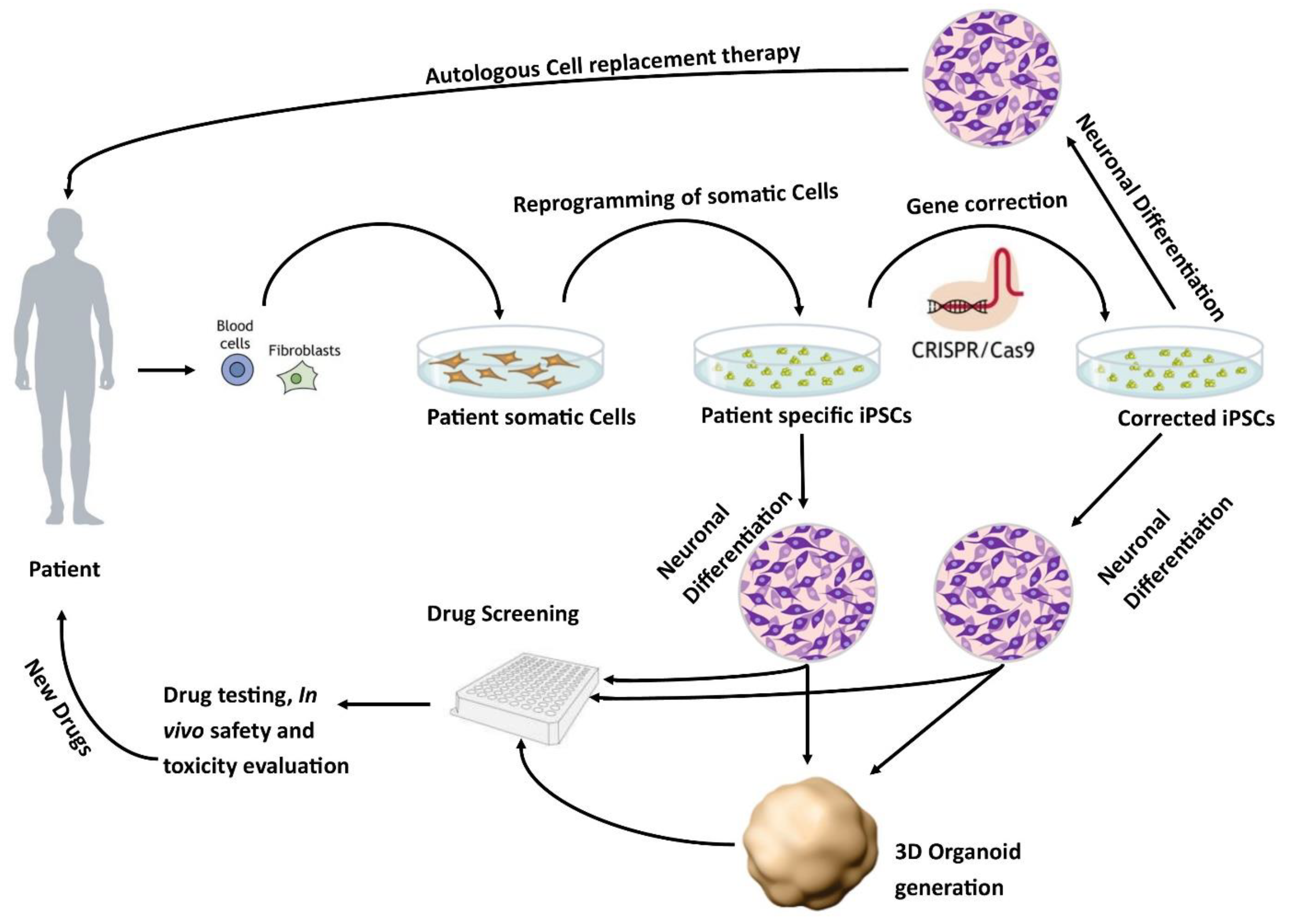
Figure 1. Patient-specific stem cell lines for cell replacement therapy and new drug development. Somatic cells from the patient’s skin or blood can be isolated and reprogrammed into iPSCs. These cells have the potential to differentiate into different neuronal populations and can be used to either further study the pathophysiology of the disease or as a drug screening system to test new potential therapies. In addition, these lines can be corrected using CRISPR/Cas9 to generate isogenic controls which provide ideal controls.
These models better recapitulate the complexity of genetic epilepsy. In recent years, stem cell research has focused on using patient-specific hiPSC-derived neurons as in vitro models of epilepsy.
2. Gene Editing Techniques for Disease Modelling and Isogenic Control
The methodology used to induce genetic variance in healthy cells for disease modelling, or to correct patient cells for isogenic control, has evolved over time. Sixteen studies (Table S1) reported the use of gene editing techniques to create isogenic controls or to study the disease-causing genetic variation in patient-derived iPSCs. Transcription Activator-Like Effector Nucleases (TALEN) was one of the first gene-editing techniques used in 2014 to generate the SCN1A mutation in human iPSCs [61][26] and was subsequently used in two more studies [48,55][27][28]. Zinc-Finger Nucleases (ZFNs) is another gene-editing technology and was used to model TSC in 2016 [35][29]. In addition, virus- or vector-based knock-out and knock-down techniques were also used in a few studies [33,42,95,106][30][31][32][33]. These methods were subsequently replaced with the more advanced CRISPR/Cas9 method. Unlike its predecessors, TALEN and ZFNs, it is precise, robust, and site-specific with fewer off-target effects [113][34]. The first use of CRISPR/Cas9 technology in epilepsy was reported in 2016 to generate a loss of function SCN1A mutation in human iPSCs to gain insight into Dravet syndrome [114][35]. This approach has been widely adopted in recent years with eight additional studies generating disease-specific neurons and isogenic controls using CRISPR/Cas9 [37,44,68,70,74,97,112,114][35][36][37][38][39][40][41][42].
3. Epilepsy Patient-Specific iPSCs Derived Disease Models
Dravet syndrome was the most commonly studied [60[26][35][38][43][44][45][46][47][48][49],61,62,63,64,65,67,68,69,114], followed by tuberous sclerosis [35[29][39][50][51][52],38,70,71,72], focal cortical dysplasia [82,83[53][54][55][56][57],84,85,104], and a few rare epilepsy syndromes [44,45,46,47,53,73,74,76,77,79,80,81,87,115][37][40][58][59][60][61][62][63][64][65][66][67][68][69]. These models have the potential to provide useful information about the involvement of a particular gene in disease progression and its anticonvulsant response.
The first in vitro model from a Dravet patient carrying a mutation in the SCN1A gene was generated in 2013. The findings from that study showed that the loss of function in GABAergic inhibition appears to be the main driver in epileptogenesis [62][44]. Since then, there have been several studies assessing various mutations in the SCN1A gene [48,54,55,56,60,61,62,63,64,65,66,67,68,114][26][27][28][35][38][43][44][45][46][47][48][70][71][72]. Neurons derived from these patients exhibit increased sodium currents and hyperexcitability [64][46], which can be alleviated by treatment with phenytoin [60][43]. In another study, neurons derived from two patients with Dravet syndrome demonstrated that genetic alterations of SCN1A differentially impacted electrophysiological impairment. The degree of impairment corresponded with the symptomatic severity of the donor from which the iPSCs were derived [63][45]. Recently, another patient-specific iPSCs-derived neuronal study generated from individuals with SCN1A mutation indicated an imbalance in excitation and inhibition that leads to hyperactivity in the neural network. This study used homozygous and isogenic controls to show the hyperexcitability in the generated neurons [68][38]. These studies indicated that neurons could recapitulate the neuronal pathophysiology and could potentially be used for screening drugs for personalised therapies [60,64][43][46].
The most commonly studied mTORopathies were tuberous sclerosis and focal cortical dysplasia (FCD). A study using neurons derived from a patient with TSC2 mutation reported hyperactivation of mTORC1 pathway [35][29]. In this model, pharmacological inhibition of mTORC1 with rapamycin reverses developmental abnormalities and synaptic dysfunction during independent developmental stages [35][29]. In another study, neuronal progenitor cells (NPCs) generated from a patient carrying a heterozygous TSC2 mutation exhibited disrupted neuronal development, potentially contributing to the disease neuropathology. Moreover, NPCs also exhibited activation of mTORC1 downstream signalling and attenuation of PI3K/AKT signalling upstream of TSC [72][52]. More recently, NPCs generated from the patient carrying TSC germline nonsense mutation in exon 15 of TSC1 showed the influence of TSC1 mutation in the early neurodevelopmental phenotypes, signalling, and gene expression in NPCs compared to the genetically matched wild-type cells [70][39]. In 2019, Sundberg et al showed that loss of one allele of TSC2 is sufficient to cause some morphological and physiological changes, elevated phosphorylation, and hyperexcitability of mTORC1 in human neurons, but biallelic mutations in TSC2 are necessary to induce gene expression dysregulation seen in cortical tubers. They also found that treatment of TSC2 patient-specific iPSCs-derived neurons with rapamycin reduced neuronal activity and partially reversed gene expression abnormalities [71][51]. In 2020, Alsaqati used commercially available TSC2 (loss of function mutation) patient-derived iPSCs and reported that the dysfunctional neuronal network behaviour in the differentiated neurons could not be rescued with rapamycin treatment [57][73]. The difference in response in these two studies is because of two different iPSCs samples carrying different mutations in the TSC2 gene [57,71][51][73].
Five studies assessed the FCD-related cortical malformation by generating iPSCs from patients with mutations in genes involved in regulating the mTOR pathway. In 2017, Marinowic et al described the generation of iPSC-based cellular models of refractory epilepsy from the fibroblasts of two refractory epilepsy patients with FCD type IIb, one a 45-year-old male and the other 12-years-old female [84][55]. Then in 2020, Marinowic published another study using these cells investigating the differences in the migration potential and the expression of genes for cell proliferation, adhesion, and apoptosis. The main finding of the study was that the gene expression was different between the neurons generated from the adult male compared to the child. They concluded that differences in the migration potential of adult cells, and differences in the expression of genes related to the fundamental brain development processes, might be associated with cortical alteration in the two patients with FCD IIb [85][56]. In 2018, Majolo et al studied the Notch signalling pathway, a pathway involved in cortical development to regulate neuronal differentiation, self-renewal, survival, and neuronal plasticity, using the iPSCs from FCD IIb patients. The study assess the expression of genes involved in Notch signalling and showed that, during embryonic neurogenesis, the neural precursor cells of FCD type IIb individuals exhibited an increase in HEY1 and NOTCH1 genes as well as a decrease in the expression of HES1 and PAX5 genes, compared to the cells from control subjects [82][53]. In the subsequent study, Majolo et al studied the migration and synaptic aspects of neurons generated using the iPSCs derived from patients with FCD type IIb. Using real-time PCR, the study presented the expression of most of the synaptic and ion channels genes ASCL1, DCX, DLG4, FGF2, NEFL, NEUROD2, NEUROD6, NRCAM, and STX1A in different groups; fibroblasts, iPSCs, differentiated neurons, and brain tissues [83][54]. This study suggested that the cells derived from FCD patients may have more sensitivity to stimuli resulting in altered cell survival, apoptosis, migration, and morphological development. In 2021, Klofas et al published a study using the FCD patient-derived neurons carrying a heterozygous loss-of-function mutation in the DEPDC5 gene and reported hyperactivation of mTORC1 and enlarged cell somas that were rescued with the inhibition of mTORC1. This study also reported that cell starvation leads to hyperactivation of the mTOR pathway [104][57] but the exact mechanism is still unclear. None of these FCD studies have performed electrophysiological functional analysis of the generated neurons.
References
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18024.
- Perucca, P.; Perucca, E. Identifying mutations in epilepsy genes: Impact on treatment selection. Epilepsy Res. 2019, 152, 18–30.
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25.
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53.
- Claes, L.R.; Deprez, L.; Suls, A.; Baets, J.; Smets, K.; Van Dyck, T.; Deconinck, T.; Jordanova, A.; De Jonghe, P. The SCN1A variant database: A novel research and diagnostic tool. Hum. Mutat. 2009, 30, E904–E920.
- Steinlein, O.K.; Mulley, J.C.; Propping, P.; Wallace, R.H.; Phillips, H.A.; Sutherland, G.R.; Scheffer, I.E.; Berkovic, S.F. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 1995, 11, 201.
- Baybis, M.; Yu, J.; Lee, A.; Golden, J.A.; Weiner, H.; McKhann, G.; Aronica, E.; Crino, P.B. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 478–487.
- Baulac, S.; Ishida, S.; Marsan, E.; Miquel, C.; Biraben, A.; Nguyen, D.K.; Nordli, D.; Cossette, P.; Nguyen, S.; Lambrecq, V. Familial focal epilepsy with focal cortical dysplasia due to DEPDC 5 mutations. Ann. Neurol. 2015, 77, 675–683.
- D’Gama, A.M.; Geng, Y.; Couto, J.A.; Martin, B.; Boyle, E.A.; LaCoursiere, C.M.; Hossain, A.; Hatem, N.E.; Barry, B.J.; Kwiatkowski, D.J. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann. Neurol. 2015, 77, 720–725.
- Lim, J.S.; Kim, W.; Kang, H.-C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.-W.; Kim, S.; Kim, H.M.; Kim, J.A. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 2015, 21, 395.
- Marsan, E.; Baulac, S. Mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 2018, 44, 6–17.
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.H. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e87.
- Scerri, T.; Riseley, J.R.; Gillies, G.; Pope, K.; Burgess, R.; Mandelstam, S.A.; Dibbens, L.; Chow, C.W.; Maixner, W.; Harvey, A.S. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC 5. Ann. Clin. Transl. Neurol. 2015, 2, 575–580.
- Sim, J.C.; Scerri, T.; Fanjul-Fernández, M.; Riseley, J.R.; Gillies, G.; Pope, K.; Van Roozendaal, H.; Heng, J.I.; Mandelstam, S.A.; McGillivray, G. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann. Neurol. 2016, 79, 132–137.
- Van Kranenburg, M.; Hoogeveen-Westerveld, M.; Nellist, M. Preliminary Functional Assessment and Classification of DEPDC 5 Variants Associated with Focal Epilepsy. Hum. Mutat. 2015, 36, 200–209.
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319.
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077.
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-resistant epilepsy. N. Engl. J. Med. 2011, 365, 919–926.
- Wiebe, S.; Blume, W.T.; Girvin, J.P.; Eliasziw, M. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N. Engl. J. Med. 2001, 345, 311–318.
- Dragunow, M. The adult human brain in preclinical drug development. Nat. Rev. Drug Discov. 2008, 7, 659.
- Grainger, A.I.; King, M.C.; Nagel, D.A.; Parri, H.R.; Coleman, M.D.; Hill, E.J. In vitro models for seizure-liability testing using induced pluripotent stem cells. Front. Neurosci. 2018, 12, 590.
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno, L.S., Jr.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705.
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130.
- Easter, A.; Bell, M.E.; Damewood, J.R., Jr.; Redfern, W.S.; Valentin, J.-P.; Winter, M.J.; Fonck, C.; Bialecki, R.A. Approaches to seizure risk assessment in preclinical drug discovery. Drug Discov. Today 2009, 14, 876–884.
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834.
- Chen, W.; Liu, J.; Zhang, L.; Xu, H.; Guo, X.; Deng, S.; Liu, L.; Yu, D.; Chen, Y.; Li, Z. Generation of the SCN1A epilepsy mutation in hiPS cells using the TALEN technique. Sci. Rep. 2014, 4, 5404.
- Zhao, H.; He, L.; Li, S.; Huang, H.; Tang, F.; Han, X.; Lin, Z.; Tian, C.; Huang, R.; Zhou, P.; et al. Generation of corrected-hiPSC (USTCi001-A-1) from epilepsy patient iPSCs using TALEN-mediated editing of the SCN1A gene. Stem Cell Res. 2020, 46, 101864.
- Tanaka, Y.; Sone, T.; Higurashi, N.; Sakuma, T.; Suzuki, S.; Ishikawa, M.; Yamamoto, T.; Mitsui, J.; Tsuji, H.; Okano, H. Generation of D1-1 TALEN isogenic control cell line from Dravet syndrome patient iPSCs using TALEN-mediated editing of the SCN1A gene. Stem Cell Res. 2018, 28, 100–104.
- Costa, V.; Aigner, S.; Vukcevic, M.; Sauter, E.; Behr, K.; Ebeling, M.; Dunkley, T.; Friedlein, A.; Zoffmann, S.; Meyer, C.A. mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 2016, 15, 86–95.
- Meganathan, K.; Lewis, E.M.A.; Gontarz, P.; Liu, S.; Stanley, E.G.; Elefanty, A.G.; Huettner, J.E.; Zhang, B.; Kroll, K.L. Regulatory networks specifying cortical interneurons from human embryonic stem cells reveal roles for CHD2 in interneuron development. Proc. Natl. Acad. Sci. USA 2017, 114, E11180–E11189.
- Fedele, D.E.; Koch, P.; Scheurer, L.; Simpson, E.M.; Mohler, H.; Brustle, O.; Boison, D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci. Lett. 2004, 370, 160–165.
- Patzke, C.; Südhof, T.C. The conditional KO approach: Cre/Lox technology in human neurons. Rare Dis. 2016, 4, 3560–3571.
- Nebel, R.; Zhao, D.; Pedrosa, E.; Kirschen, J.; Lachman, H.; Zheng, D.; Abrahams, B. Reduced CYFIP1 in iPSC Derived Human Neural Progenitors Results in Donor Specific Dysregulation of Schizophrenia and Epilepsy Genes. Neuropsychopharmacology 2015, 40, S379.
- Heidenreich, M.; Zhang, F. Applications of CRISPR–Cas systems in neuroscience. Nat. Rev. Neurosci. 2016, 17, 36.
- Liu, J.; Gao, C.; Chen, W.; Ma, W.; Li, X.; Shi, Y.; Zhang, H.; Zhang, L.; Long, Y.; Xu, H. CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: Mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl. Psychiatry 2016, 6, e703.
- Zhu, W.; Zhang, B.; Li, M.; Mo, F.; Mi, T.; Wu, Y.; Teng, Z.; Zhou, Q.; Li, W.; Hu, B. Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome. Nat. Commun. 2019, 10, 928.
- Malerba, N.; Benzoni, P.; Squeo, G.M.; Milanesi, R.; Giannetti, F.; Sadleir, L.G.; Poke, G.; Augello, B.; Croce, A.I.; Barbuti, A.; et al. Generation of the induced human pluripotent stem cell lines CSSi009-A from a patient with a GNB5 pathogenic variant, and CSSi010-A from a CRISPR/Cas9 engineered GNB5 knock-out human cell line. Stem Cell Res. 2019, 40, 101547.
- Xie, Y.; Ng, N.N.; Safrina, O.S.; Ramos, C.M.; Ess, K.C.; Schwartz, P.H.; Smith, M.A.; O’Dowd, D.K. Comparisons of dual isogenic human iPSC pairs identify functional alterations directly caused by an epilepsy associated SCN1A mutation. Neurobiol. Dis. 2020, 134, 104627.
- Martin, P.; Wagh, V.; Reis, S.A.; Erdin, S.; Beauchamp, R.L.; Shaikh, G.; Talkowski, M.; Thiele, E.; Sheridan, S.D.; Haggarty, S.J. TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol. Autism 2020, 11, 2.
- Burnight, E.R.; Bohrer, L.R.; Giacalone, J.C.; Klaahsen, D.L.; Daggett, H.T.; East, J.S.; Madumba, R.A.; Worthington, K.S.; Mullins, R.F.; Stone, E.M. CRISPR-Cas9-Mediated correction of the 1.02 kb common deletion in CLN3 in induced pluripotent stem cells from patients with batten disease. CRISPR J. 2018, 1, 75–87.
- Simkin, D.; Marshall, K.A.; Vanoye, C.G.; Desai, R.R.; Bustos, B.I.; Piyevsky, B.N.; Ortega, J.A.; Forrest, M.; Robertson, G.L.; Penzes, P. Dyshomeostatic modulation of Ca2+-activated K+ channels in a human neuronal model of KCNQ2 encephalopathy. Elife 2021, 10, e64434.
- Fatima, A.; Schuster, J.; Akram, T.; Sobol, M.; Hoeber, J.; Dahl, N. Generation of a human Neurochondrin deficient iPSC line KICRi002-A-3 using CRISPR/Cas9. Stem Cell Res. 2020, 44, 101758.
- Jiao, J.; Yang, Y.; Shi, Y.; Chen, J.; Gao, R.; Fan, Y.; Yao, H.; Liao, W.; Sun, X.F.; Gao, S. Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum. Mol. Genet. 2013, 22, 4241–4252.
- Higurashi, N.; Uchida, T.; Lossin, C.; Misumi, Y.; Okada, Y.; Akamatsu, W.; Imaizumi, Y.; Zhang, B.; Nabeshima, K.; Mori, M.X. A human Dravet syndrome model from patient induced pluripotent stem cells. Mol. Brain 2013, 6, 19.
- Kim, H.W.; Quan, Z.; Kim, Y.-B.; Cheong, E.; Kim, H.D.; Cho, M.; Jang, J.; Yoo, Y.R.; Lee, J.S.; Kim, J.H. Differential effects on sodium current impairments by distinct SCN1A mutations in GABAergic neurons derived from Dravet syndrome patients. Brain Dev. 2018, 40, 287–298.
- Liu, Y.; Lopez-Santiago, L.F.; Yuan, Y.; Jones, J.M.; Zhang, H.; O’Malley, H.A.; Patino, G.A.; O’Brien, J.E.; Rusconi, R.; Gupta, A. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol. 2013, 74, 128–139.
- Maeda, H.; Chiyonobu, T.; Yoshida, M.; Yamashita, S.; Zuiki, M.; Kidowaki, S.; Isoda, K.; Yamakawa, K.; Morimoto, M.; Nakahata, T. Establishment of isogenic iPSCs from an individual with SCN1A mutation mosaicism as a model for investigating neurocognitive impairment in Dravet syndrome. J. Hum. Genet. 2016, 61, 565.
- Sun, Y.; Dolmetsch, R.E. Investigating the therapeutic mechanism of cannabidiol in a human induced pluripotent stem cell (iPSC)-based model of Dravet syndrome. Cold Spring Harb. Symp. Quant. Biol. 2018, 83, 185–191.
- Fruscione, F.; Valente, P.; Sterlini, B.; Romei, A.; Baldassari, S.; Fadda, M.; Prestigio, C.; Giansante, G.; Sartorelli, J.; Rossi, P.; et al. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain 2018, 141, 1000–1016.
- Rocktäschel, P.; Sen, A.; Cader, M.Z. High glucose concentrations mask cellular phenotypes in a stem cell model of tuberous sclerosis complex. Epilepsy Behav. 2019, 101, 106581.
- Winden, K.D.; Sundberg, M.; Yang, C.; Wafa, S.M.; Dwyer, S.; Chen, P.-F.; Buttermore, E.D.; Sahin, M. Biallelic mutations in TSC2 lead to abnormalities associated with cortical tubers in human iPSC-derived neurons. J. Neurosci. 2019, 39, 9294–9305.
- Zucco, A.J.; Dal Pozzo, V.; Afinogenova, A.; Hart, R.P.; Devinsky, O.; D’Arcangelo, G. Neural progenitors derived from tuberous sclerosis complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol. Cell. Neurosci. 2018, 92, 149–163.
- Majolo, F.; Marinowic, D.; Machado, D.; Da Costa, J.C. Notch signaling in human iPS-derived neuronal progenitor lines from Focal Cortical Dysplasia patients. Int. J. Dev. Neurosci. 2018, 69, 112–118.
- Majolo, F.; Marinowic, D.R.; Palmini, A.L.F.; DaCosta, J.C.; Machado, D.C. Migration and synaptic aspects of neurons derived from human induced pluripotent stem cells from patients with focal cortical dysplasia II. Neuroscience 2019, 408, 81–90.
- Marinowic, D.R.; Majolo, F.; Sebben, A.D.; Da Silva, V.D.; Lopes, T.G.; Paglioli, E.; Palmini, A.; Machado, D.C.; Da Costa, J.C. Induced pluripotent stem cells from patients with focal cortical dysplasia and refractory epilepsy. Mol. Med. Rep. 2017, 15, 2049–2056.
- Marinowic, D.R.; Majolo, F.; Zanirati, G.G.; Plentz, I.; Neto, E.P.; Palmini, A.L.F.; Machado, D.C.; Da Costa, J.C. Analysis of genes involved in cell proliferation, adhesion, and control of apoptosis during embryonic neurogenesis in Induced Pluripotent Stem Cells (iPSCs) from patients with Focal Cortical Dysplasia. Brain Res. Bull. 2020, 155, 112–118.
- Klofas, L.K.; Short, B.P.; Snow, J.P.; Sinnaeve, J.; Rushing, G.V.; Westlake, G.; Weinstein, W.; Ihrie, R.A.; Ess, K.C.; Carson, R.P. DEPDC5 haploinsufficiency drives increased mTORC1 signaling and abnormal morphology in human iPSC-derived cortical neurons. Neurobiol. Dis. 2020, 143, 104975.
- Schwarz, N.; Uysal, B.; Rosa, F.; Löffler, H.; Mau-Holzmann, U.A.; Liebau, S.; Lerche, H. Generation of an induced pluripotent stem cell (iPSC) line from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p. Leu328Val) mutation. Stem Cell Res. 2018, 33, 6–9.
- Sun, C.; Yang, M.; Qin, F.; Guo, R.; Liang, S.; Hu, H. Generation of an induced pluripotent stem cell line SYSUi-003-A from a child with epilepsy carrying GRIN2A mutation. Stem Cell Res. 2020, 43, 101706.
- Zhang, B.; Wang, Y.; Peng, J.; Hao, Y.; Guan, Y. Generation of a human induced pluripotent stem cell line from an epilepsy patient carrying mutations in the PIK3R2 gene. Stem Cell Res. 2020, 44, 101711.
- Tan, G.W.; Kondo, T.; Murakami, N.; Imamura, K.; Enami, T.; Tsukita, K.; Shibukawa, R.; Funayama, M.; Matsumoto, R.; Ikeda, A. Induced pluripotent stem cells derived from an autosomal dominant lateral temporal epilepsy (ADLTE) patient carrying S473L mutation in leucine-rich glioma inactivated 1 (LGI1). Stem Cell Res. 2017, 24, 12–15.
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 2017, 20, 435–449.e4.
- Chou, S.-J.; Tseng, W.-L.; Chen, C.-T.; Lai, Y.-F.; Chien, C.-S.; Chang, Y.-L.; Lee, H.-C.; Wei, Y.-H.; Chiou, S.-H. Impaired ROS scavenging system in human induced pluripotent stem cells generated from patients with MERRF syndrome. Sci. Rep. 2016, 6, 23661.
- Fink, J.J.; Robinson, T.M.; Germain, N.D.; Sirois, C.L.; Bolduc, K.A.; Ward, A.J.; Rigo, F.; Chamberlain, S.J.; Levine, E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017, 8, 15038.
- Gillentine, M.A.; Yin, J.; Bajic, A.; Zhang, P.; Cummock, S.; Kim, J.J.; Schaaf, C.P. Functional consequences of CHRNA7 copy-number alterations in induced pluripotent stem cells and neural progenitor cells. Am. J. Hum. Genet. 2017, 101, 874–887.
- Guemez-Gamboa, A.; Çağlayan, A.O.; Stanley, V.; Gregor, A.; Zaki, M.S.; Saleem, S.N.; Musaev, D.; McEvoy-Venneri, J.; Belandres, D.; Akizu, N. Loss of Protocadherin-12 L eads to D iencephalic-M esencephalic J unction D ysplasia S yndrome. Ann. Neurol. 2018, 84, 638–647.
- Homan, C.C.; Pederson, S.; To, T.H.; Tan, C.; Piltz, S.; Corbett, M.A.; Wolvetang, E.; Thomas, P.Q.; Jolly, L.A.; Gecz, J. PCDH19 regulation of neural progenitor cell differentiation suggests asynchrony of neurogenesis as a mechanism contributing to PCDH19 Girls Clustering Epilepsy. Neurobiol. Dis. 2018, 116, 106–119.
- Shahsavani, M.; Pronk, R.J.; Falk, R.; Lam, M.; Moslem, M.; Linker, S.B.; Salma, J.; Day, K.; Schuster, J.; Anderlid, B.M.; et al. An in vitro model of lissencephaly: Expanding the role of DCX during neurogenesis. Mol. Psychiatry 2018, 23, 1674–1684.
- Gamboa, A.G.; Rakotomamonjy, J.; Rylaarsdaam, L.; Thomas, D. Loss of PCDH12 causes cell migration and differentiation defects in human embryonic stem cell-derived neuroprogenitors. J. Neurochem. 2019, 150, 191.
- Tanaka, Y.; Higurashi, N.; Shirasu, N.; Yasunaga, S.i.; Moreira, K.M.; Okano, H.; Hirose, S. Establishment of a human induced stem cell line (FUi002-A) from Dravet syndrome patient carrying heterozygous R1525X mutation in SCN1A gene. Stem Cell Res. 2018, 31, 11–15.
- Zhao, H.; Li, S.; He, L.; Han, X.; Huang, H.; Tang, F.; Lin, Z.; Deng, S.; Tian, C.; Huang, R. Generation of iPSC line (USTCi001-A) from human skin fibroblasts of a patient with epilepsy. Stem Cell Res. 2020, 45, 101785.
- Schuster, J.; Fatima, A.; Sobol, M.; Norradin, F.H.; Laan, L.; Dahl, N. Generation of three human induced pluripotent stem cell (iPSC) lines from three patients with Dravet syndrome carrying distinct SCN1A gene mutations. Stem Cell Res. 2019, 39, 101523.
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol. Autism 2020, 11, 80.
More