T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive subtype of hematological malignancy characterized by its high heterogeneity and potentially life-threatening clinical features. It has shown the indispensable effects of leukemia-initiating cells (LICs) and leukemic niches on T-ALL initiation and progression. These milestones greatly facilitate precision medicine by interfering with the pathways that are associated with LICs and leukemic niches or by targeting themselves directly. Most of these novel agents, either alone or in combination with conventional chemotherapy, have shown promising preclinical results, facilitating them to be further evaluated under clinical trials.
- T-cell acute lymphoblastic leukemia
- leukemia-initiating cell
- microenvironment
- leukemic niche
1. Introduction
2. LICs in T-ALL
LICs, also termed leukemia stem cells (LSCs), were first identified by Dick and his colleagues in studies on acute myeloid leukemia (AML) [3][4], characterized by their self-renewal capability and potential to differentiate into leukemic blasts [4][5][5,6]. Although LICs and LSCs are used interchangeably for AML, their concepts are not necessarily the same [6][7]. The LICs more appropriately denote the leukemia cells of origin, whereas the LSCs refer to a distinct subpopulation with the capacity for self-renewal and long-term clonal maintenance at a later stage [5][6][6,7]. In this context, the LICs are at the apex of the leukemic hierarchy, whereas the LSCs represent cells that can be prospectively isolated from the remainder of the cancer cells based on specific cell surface markers. However, in some cancers such as T-ALL, it is not possible to distinguish LSCs from non-LSCs due to the ill-defined immunophenotypes. In this regard, Dick and others have defined such cells as LICs by their ability to (i) generate leukemia in transplanted xenografts, (ii) self-renew upon serial passages in xenografts, and (iii) give rise to daughter cells with proliferative capacity but that are unable to maintain the tumor clone after serial passages [5][6]. Nonetheless, the definition of LICs in T-ALL are not well characterized. In most instances, the T-ALL LICs (T-LICs) and LSCs are still used interchangeably, whereas in some studies, the T-LICs only refer to the transplanted cells with leukemia-initiating capacity. As such, a unified term of T-LICs refers to all cells with leukemia-initiating potential and/or LSC capacity.2.1. T-LICs in Mouse Models
T-LICs were mostly studied in genetically engineered mouse models of T-ALL, among which, the T-cell acute lymphocytic 1 (TAL) basic helix-loop-helix (bHLH) transcription factor 1 (Tal1)-induced mouse model stands out, since 28% of the Tal1 transgenic mice develop leukemia [7][8]. Additionally, co-expression of Tal1 with LIM domain only 1 (Lmo1) or Lmo2 not only accelerates T-ALL onset and progression [8][9][9,10] but also provides a favorable context for the acquisition of activating mutations of notch receptor 1 (NOTCH1) and the emergence of T-LICs [10][11]. Thus, the Tal1-Lmo1/2 transgenic mouse is commonly utilized as a model for characterizing T-LICs [10][11][11,12]. Moreover, the retroviral or transgenic mouse models of Notch1-induced T-ALL [12][13][14][15][16][13,14,15,16,17], KRAS proto-oncogene (Kras)G12D-induced T-ALL [12][17][13,18], and phosphatase and tensin homolog (Pten)-null T-ALL [18][19][19,20] are also used for identification of T-LICs in terms of different T-ALL-causing mutations. Of note, the genes that encode TAL1 and LMO1/2 are recurring targets of chromosomal translocation [20][21], and the activating mutations of NOTCH1 were identified in more than 60% of human T-ALL cases [21][22][22,23]. Interestingly, NOTCH1 was revealed as a key regulator of human T-LIC activity, since inhibition of the NOTCH1 pathway by γ-secretase inhibitors (GSIs) abolishes T-LIC activity in xenografts and mouse models [11][23][24][12,24,25]. However, the gain-of-function mutations in NOTCH1 can initiate T-ALL in mouse models but have weak leukemogenic strength, implying that additional cooperating events are required [12][13]. Indeed, NOTCH1 modulates T-LIC activity by cooperating with oncogenic TAL1-LMO1/2 transcription factors [10][11], KRAS [12][17][13,18], runt-related transcription factor (RUNX)-mediated regulation of protein kinase C theta (PKCθ) and reactive oxygen species (ROS) [13][14], MYC proto-oncogene (MYC) [15][16], cyclin dependent kinase 6 (CDK6) [16][17], or CD44 [25][26] (Figure 1A).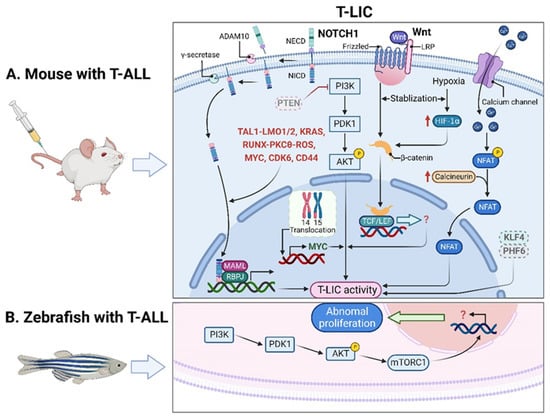
2.2. T-LICs in Zebrafish Models
2.3. Therapies Targeting T-LICs
3. Leukemic Niches in T-ALL
3.1. Effects of Leukemic Niches on T-ALL Cells
3.1.1. CXC Chemokine Ligand 12 (CXCL12)/CXC Chemokine Receptor 4 (CXCR4) Signaling
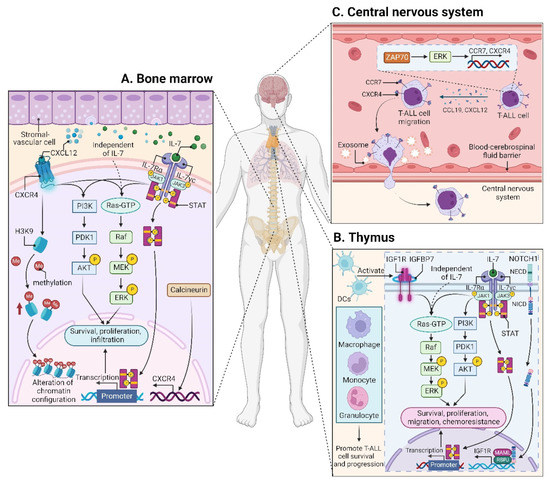
CXCL12 (also termed stromal cell derived factor-1, SDF-1) is a chemotactic factor, and its receptor CXCR4 is a highly conserved G-protein-coupled seven transmembrane receptor. Since stromal-vascular BM niches secrete abundant CXCL12, whereas CXCR4 is primarily expressed on HSCs and malignant cells, the CXCL12/CXCR4 axis becomes a key pathway for both normal hematopoiesis and cancer cell homing to the BM [48][49][50][51][62,63,64,65]. Indeed, T-ALL cells express high surface levels of CXCR4 in a calcineurin-dependent manner, and CXCR4 silencing impairs migration and survival of leukemic cells, as well as T-LIC activity [52][66]. Likewise, CXCL12 deletion from vascular endothelial niches impedes T-ALL development [53][67]. Mechanistically, the interaction between CXCL12 and CXCR4 results in activation of many survival pathways including PI3K-AKT and mitogen-activated protein kinase (MAPK) signaling cascades in T-ALL cells [54][68] (Figure 2A). The CXCL12/CXCR4 signaling also mediates enhanced extramedullary infiltration and dissemination [55][56][69,70]. Therefore, the activation of CXCL12/CXCR4 signaling creates a sanctuary microenvironment for T-ALL cells and confers resistance to conventional therapies [54][57][68,71]. As such, inhibition of either the CXCL12/CXCR4 interaction or its downstream signaling is of therapeutic benefit in patients with chemoresistant T-ALL [58][72].
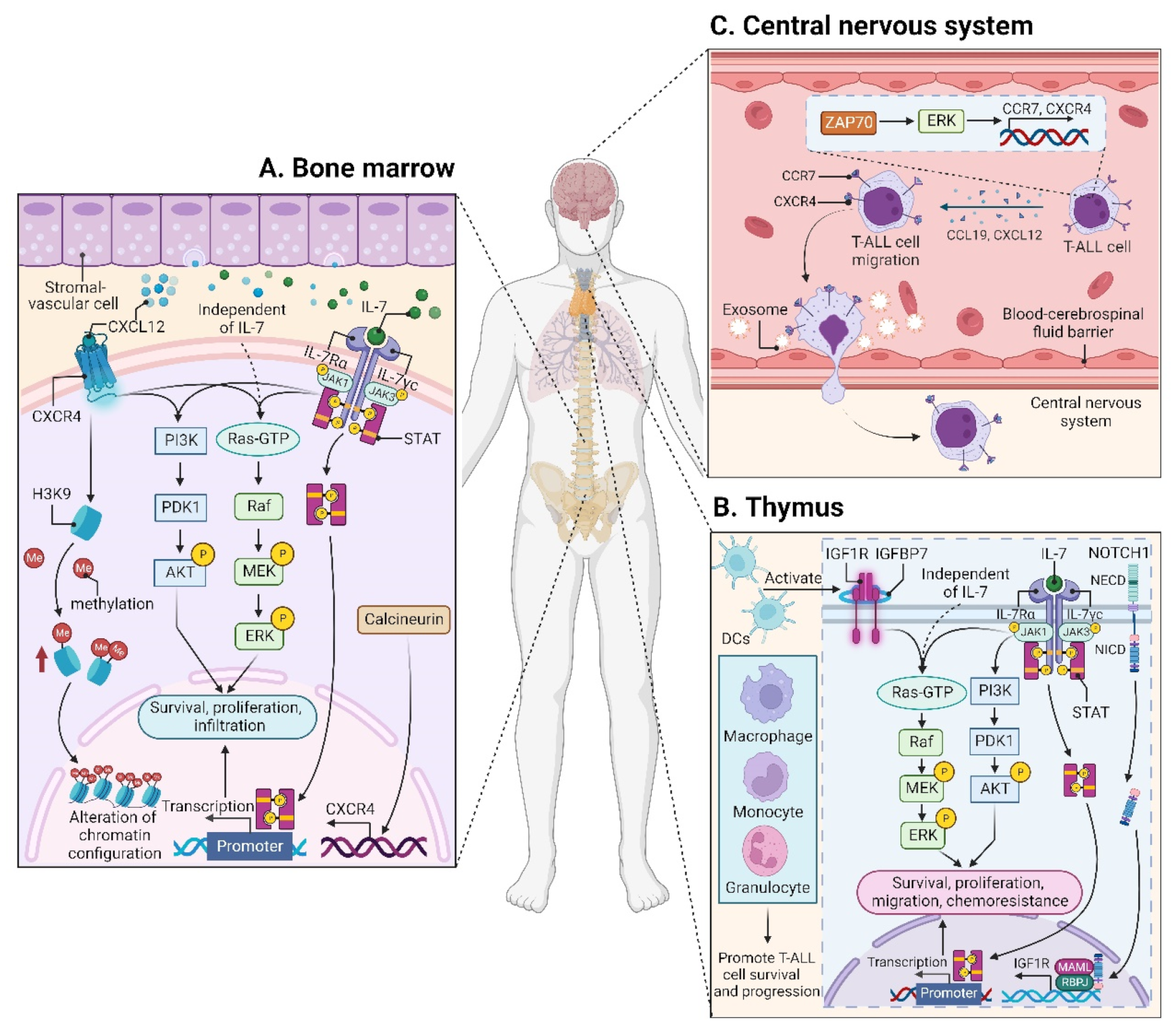
3.1.2. Insulin-like Growth Factor 1 (IGF1)/IGF1 Receptor (IGF1R) Signaling
3.1.3. Interleukin (IL7)/IL7 Receptor (IL7R) Signaling
IL7 is produced by stromal cells in the BM and thymus. It binds to IL7R, a heterodimer composed of an IL7Rα chain and a γc chain [61][101]. The IL7/IL7R signaling is critical to the survival and proliferation of thymocytes, and the tight regulation of IL7 and IL7Rα is essential for T-cell development. Hence, it is not surprising that aberrant expression or dysfunction of the IL7/IL7R axis contributes to the pathogenesis of T-ALL [62][63][102,103]. Several lines of evidence have demonstrated that the somatic gain-of-function mutation in IL7Ra exon 6 is a known driver of T-ALL, occurring in roughly 9% of pediatric and 12% of adult T-ALL cases [64][65][66][67][68][69][104,105,106,107,108,109]. Mechanistically, IL7Ra mutations induce activation of Janus kinase (JAK)-signal transducer and activator of transcription (STAT), PI3K-AKT, and MAPK kinase (MEK)-extracellular regulated protein kinase (ERK) pathways [64][70][71][76,104,110]. Overexpression of wild-type IL7Rα also promotes T-cell tumorigenesis via JAK-STAT, PI3K-AKT, and cell-cycle-related signaling, even in the absence of IL7Rα mutational activation [72][111]. Moreover, high levels of IL7Rα expression lead to increased T-LIC activity mediated by the activating mutations in NOTCH1 [73][112] or the loss-of-function mutations in DYNAMIN2 [74][113]. These findings pinpoint that not only mutational activation of IL7Rα but also high IL7Rα levels are oncogenic in T-ALL (Figure 2B). In addition to IL7Rα, mutations in other components of the IL7R-mediated signaling cascade (e.g., JAK1, JAK3, STAT5) were also identified as critical drivers for T-ALL [66][68][75][76][77][78][106,108,114,115,116,117]. These mutations and the resulting aberrant signaling provide a therapeutic window of opportunity [68][108].3.1.4. CC Chemokine Ligand 19 (CCL19)/CC Chemokine Receptor 7 (CCR7) Signaling
Patients with T-ALL are at a high risk of CNS relapse [20][21], which requires intensified intrathecal chemotherapy and systemic administration of CNS-penetrating therapeutics. Unfortunately, the mechanisms accounting for the infiltration of T-ALL cells into CNS remain incompletely understood, and little is known about their crosstalk. The CCL19/CCR7 signaling was identified as a key CNS entry signal, which is both necessary and sufficient for T-ALL cells targeting the CNS. Silencing either CCR7 or CCL19 specifically inhibits CNS infiltration [79][118]. Furthermore, the BM infiltration constitutes a prerequisite for CNS pathology in T-ALL via the CXCR4-mediated signaling [56][70]. In this sense, CNS invasion by T-ALL cells is very likely attributed to the BM microenvironmental alterations. Interestingly, both CCR7 and CXCR4 are upregulated by zeta-chain-associated protein kinase 70 (ZAP70) via the activation of ERK signaling, and high expression of ZAP70-CCR7 confers an increased risk for CNS involvement in T-ALL patients [80][119] (Figure 2C). Therefore, therapeutic monoclonal antibodies (mAbs) targeting CCR7 not only display a strong in vitro complement-dependent cytotoxicity (CDC) and an in vivo anti-tumor activity, but also show efficacy in eradicating leukemic cells from LN and CNS [81][120]. In addition to the CCR7-mediated CNS infiltration, the exosomes isolated from a T-ALL cell line P12 but not the B-cell acute lymphoblastic leukemia (B-ALL) cell lines facilitate CNS invasion across the blood–cerebrospinal fluid (CSF) barrier without disrupting the barrier integrity [82][121] (Figure 2C), highlighting the contribution of T-ALL-derived exosomes to CNS infiltration, although further studies are needed to clarify the molecular mechanisms.3.2. Microenvironmental Alterations
3.2.1. BM Microenvironment
Modification of the BM microenvironment by leukemia cells was well-characterized in B-ALL and AML [83][84][85][126,127,128], whereas the research on the alterations of the BM microenvironment by T-ALL cells is very limited. In 2016, an elegant work reported that an accumulated T-ALL burden within the BM leads to rapid and selective remodeling of the endosteal space, resulting in a complete loss of mature osteoblasts and impairment of normal HSCs [42][56].