The von Willebrand factor (vWF) is a plasma protein that mediates platelet adhesion and leukocyte recruitment to vascular injury sites and carries coagulation factor VIII, a building block of the intrinsic pathway of coagulation. A steep rise in shear rates, which may occur at sites of arterial stenosis and injury, is crucial for unfolding and activation of vWF. The hemostatic activity of vWF is counterbalanced by ADAMTS-13, a vWF-cleaving protease. The presence of ultra-large multimers of vWF in the bloodstream is associated with spontaneous thrombosis, whereas its deficiency leads to bleeding.
- platelet adhesion
- leukocyte recruitment
- von Willebrand factor
- ADAMTS-13
- shear stress
1. Introduction
The von Willebrand Factor (vWF) is a large multimeric glycoprotein, present in blood plasma, endothelial cells, megakaryocytes, and platelets. It plays a major role in hemostasis, mediating platelet adhesion to vascular injury sites. It also binds and protects coagulation factor VIII (FVIII) from degradation[1] [1]. Recent studies showed that vWF is also involved in inflammation, linking thrombosis and inflammation. Inflammation can provoke thrombosis through the vWF-dependent pathway, which includes endothelial activation, the secretion of vWF into the bloodstream, vWF activation and interaction with platelets, and subsequent platelet adhesion to a vessel wall [2][3][4][2,3,4]. vWF multimers and platelets, adhered to injured and activated endothelium, might serve as sites of leukocyte recruitment. Altogether, this predisposes to the propagation of the inflammatory process. vWF is considered a risk factor of arterial thrombosis and might contribute to the development of adverse events in atherosclerosis and other cardiovascular diseases[5][6][7][8] [5,6,7,8].
2. Structure and Functions of VWF in Bloodstream
vWF is mainly produced in endothelial cells and stored in Weibel–Palade bodies—the storage granules of endothelium, consisting of densely packed vWF multimers and P-selectin[9] [9]. A small amount of total vWF is produced in megakaryocytes and stored in the α-granules of platelets. The mature vWF monomeric molecules form dimers through C-terminal disulphide bonds. Dimers compose the basic vWF repeating structure, with a molecular mass of approximately 500 kDa[10] [10]. Dimers are then polymerized into ultra-large multimers through N-terminal disulphide bonds, with the size of stored multimers ranging from 20 to 100 dimers[11] [11].
vWF is constantly secreted from the Weibel–Palade bodies of endothelial cells into the bloodstream, and from the α-granules of platelets upon activation. The pool of circulating vWF consists of multimers of various sizes, ranging from a few dimers to high-molecular weight multimers (HMWM), which contain 11–20 dimers. The former basically serve as FVIII carriers, while the latter present the main hemostatically active force of vWF [11]. The more dimers that are in a vWF molecule, the more hemostatically active it is. Upon secretion, large vWF multimers come into contact with metalloproteinase ADAMTS-13, the vWF-cleaving protease. ADAMTS-13 regulates the size of circulating vWF through proteolytic cleavage of its multimers [12].
vWF contains a variety of domains, which define the characteristics of the molecule: A1, A2, A3; D assemblies: D1, D2, D’D3, D4; and the C-terminal cysteine knot (CTCK) (Figure 1) [13]. The A1 domain binds mainly glycoprotein (GP) Ib receptors of platelets (the only receptor on non-activated platelets that has pronounced affinity to vWF), and to a lesser extent, collagen types I, IV, VI, and heparin. The A2 domain presents a binding cite for ADAMTS-13. The main collagen binding sites are located on the A3 domain. The C1 domain can interact with activated platelets through the binding of the IIb/IIIa receptors. The D’D3 assembly carries FVIII. The CTCK domain dimerizes vWF. All D assemblies are involved in the assembly and disulfide linkage of vWF dimers into long tubules of the Weibel–Palade bodies.[9][13] [9,13]. vWF binds leukocytes through the interaction of the A1 domain with P-selectin glycoprotein ligand-1 (PSGL-1), the D’D3, and the A1, A2, and A3 domains with β2-integrins [14].
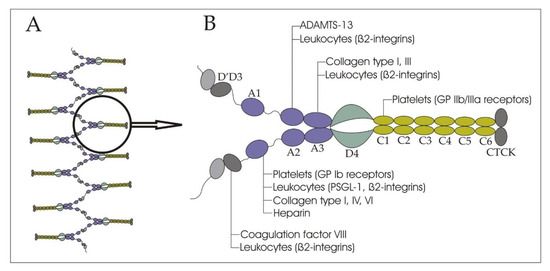
Figure 1.
A
B
vWF exists in the bloodstream in one of two conformations—globular and unfolded[15] [15]. vWF conformation depends on the shear rate of blood flow in vessels. The shear rate is the rate of change of velocity, at which one layer of fluid passes over another, and is measured in inverse, or reciprocal, seconds (s−1). This parameter is used to estimate flow of liquids in tubes, which is important for the understanding of vWF activation. Under conditions of low shear rates (e.g., in veins, where shear rates are 15–200 s−1, or in large arteries, where they are 300–800 s−1), vWF remains in a globular shape, which hides the binding sites, and does not interact with the circulating platelets[16] [16]. In the case of vessel injury and under high shear rates (e.g., in intact small arteries and arterioles, shear rates are 450–1600 s−1, but at sites of advanced atherosclerosis, shear rates might reach up to 11,000 s−1 and even higher), the vWF shape unfolds, and opens binding sites, in particular for glycoprotein (GP) Ib receptors of platelets.
A threshold value of shear rate, critical for unfolding and the activation of vWF, is estimated at 5000 s−1[15] [15]. Uniform shear rates occur in straight channels, but in pathological conditions in stenotic or injured vessels, elongational flows occur, where the molecules are subjected to shear gradients. The vasoconstriction of injured vessels and stenoses of arterial lumen by atherosclerotic plaques create two zones of elongational flow. Flow and shear rate accelerate at the inflow and decelerate at the outflow of a lesion. At the inflow, elongation occurs parallel to the flow direction. At the outflow, elongation occurs perpendicular to the flow direction because the streamlines diverge. As the adjacent shear layers have different velocities, large molecules, such as vWF multimers, are subjected to the rotational and elongational components of the flow. In the straight sections of a blood vessel, the rotational and elongational components are balanced. However, at the inflow and the outflow of a lesion, the equilibrium is disturbed. At sites of vasoconstriction and stenoses, thrombus formation is observed at the outflow zone of a lesion (Figure 2) [9]. Elongation flows are predicted to unfold vWF at rates of two orders of magnitude below the corresponding pure shear rate values [17]. vWF-dependent thrombus formation was observed at the outflow region of an in vitro model of stenosis at the inflow shear rates of 600, 1000, and 2000 s−1 [18].
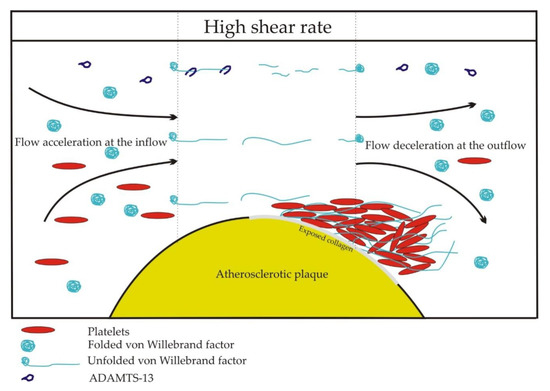
Figure 2.
Unfolded by high shear rates, the vWF threads can self-associate and form long strands and web-like structures, further stimulating platelet adhesion[19] [19]. The ability of unfolded vWF to self-associate under high-shear rates was studied in vitro in endothelialized microvessels. Smaller vessels, turns, bifurcations, flow acceleration predisposed to thickening, and elongation of vWF strands. In regions with complex flow, the vWF strands tended to form web-like structures. At the same time, at sites of low shear rates, the vWF remained in a globular conformation and no vWF strands were visible[19] [19]. Schneider et al. also reported spider web-like network formation by vWF, under high shear rates[15] [15]. Zhang et al., demonstrated that vWF association was dependent on the A2 domain under shear rate of 9600 s−1. Hence, the A2 domain might have an overlapping role, both in proteolysis and self-association of vWF[20] [20].
The role of platelet vWF in hemostasis is not clearly established, and the studies here are quite sparse. A study on the vWF-knockout mice showed that replenishing of vWF in platelets after bone marrow transplantation from wild-type mice, resulted in partial correction of bleeding time and reduction of blood loss volume[21] [21]. Recently, a study showed that chimeric mice with only platelet vWF had a bleeding time comparable to that of the vWF-knockout mice[22] [22]. Platelet vWF did not contribute to thrombus formation in a model of carotid artery injury. On the other hand, after ischemic brain injury, the mice with only platelet vWF and without plasma vWF, had a cerebral infarction size similar to the wild-type mice. Platelet vWF mediated ischemic brain injury in a GPIb-dependent mechanism and significantly contributed to intracerebral thrombosis formation[22] [22].
Upon the unfolding of vWF multimers, FVIII is exposed on D’D3 domains [1]. FVIII participates immediately in the intrinsic coagulation pathway, interacting with factor IXa as a cofactor to form the intrinsic tenase (enzyme complex that cleaves inactive coagulation factor X into active Xa). FVIII is essential for normal hemostasis—its deficiency leads to hemophilia A. vWF acts as a carrier of FVIII in circulation. In the absence of vWF, FVIII is unstable and exposed to rapid degradation. vWF significantly elongates its half-life and protects FVIII from proteolysis, eventually delivering it to sites of vessel injury[1] [1].
Unraveling, vWF faces its antagonist, ADAMTS-13[9] [9]. It is a member of the ADAMTS (A Disintegrin And Metalloprotease with ThromboSpondin type 1 repeats) family of proteolytic enzymes. ADAMTS-13 was first isolated in 2001, though the presence of Ca2+-dependent vWF-cleaving protease was predicted earlier [23]. Human ADAMTS-13 mRNA and protein synthesis is localized exclusively to hepatic stellate cells[24] [24]. The plasma levels of ADAMTS-13 show negative correlation with the plasma levels of vWF [25]. In the absence of vWF in the bloodstream, i.e., in patients with von Willebrand disease (vWD) type 3, the plasma levels of ADAMTS-13 are 30% higher than in healthy volunteers. On the contrary, after the administration of 1-deamino-8-D-arginine vasopressin (DDAVP), which stimulates the release of vWF from endothelial Weibel–Palade bodies, the plasma levels of ADAMTS-13 showed a 20% reduction in healthy volunteers[25] [25]. The negative correlation is likely to occur because of consumption in the cleavage of vWF. ADAMTS-13 circulates in the bloodstream as an active enzyme, but vWF in the globular conformation remains resistant to proteolysis. Once vWF unfolds, it exposes the binding sites for ADAMTS-13 on the A2 domain. The ensuing interaction with ADAMTS-13 leads to the cleavage of vWF multimers between tyrosine in the 1605 position and methionine in the 1606 position, which shortens the multimers and decreases the hemostatic activity of vWF [16][26][16,26].
The growing evidence shows that vWF is an important mediator of vascular inflammation [27]. A murine model of acute peritonitis showed that vWF plays an important role in leukocyte recruitment and extravasation into the site of inflammation. Notably, leukocyte extravasation requires vWF-platelet binding, as inhibition of the platelet GPIb receptors prevented vWF-mediated leukocyte extravasation [28]. A murine model of immune complex-mediated vasculitis and irritant contact dermatitis showed that the interference with vWF activity with vWF-blocking antibody led to a substantial decrease in vWF-mediated leukocyte recruitment and reduction of cutaneous inflammatory response. In this study, the vWF-blocking antibody did not interfere with the GPIb receptors [29]. Another murine model with the same pathology but with the use of the antibody targeting the A1 domain showed comparable results[30] [30]. A study comparing features of acute inflammation after focal cerebral ischemia in vWF-deficient and ADAMTS-13-deficient mice, showed that the vWF-deficient mice had a smaller injury size, reduced cytokine release and neutrophil infiltration of the injury site, compared to the ADAMTS-13-deficient mice [31]. A myocardial ischemia/reperfusion study in ADAMTS-13-knockout mice showed that infusion of recombinant ADAMTS-13 substantially reduced myocardial apoptosis, troponin-I release, and resulted in a 9-fold reduction in the number of neutrophils infiltrating the ischemic zone[32] [32].
References
-
Terraube, V.; O’Donnell, J.S.; Jenkins, P.V. Factor VIII and von Willebrand factor interaction: Biological, clinical and therapeutic importance. Haemophilia 2010, 16, 3–13.
-
Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147.
-
Kawecki, C.; Lenting, P.J.; Denis, C.V. von Willebrand factor and inflammation. J. Thromb. Haemost. 2017, 15, 1285–1294.
-
Pendu, R.; Terraube, V.; Christophe, O.D.; Gahmberg, C.G.; de Groot, P.G.; Lenting, P.J.; Denis, C.V. P-selectin glycoprotein ligand 1 and β2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood 2006, 108, 3746–3752.
-
Fan, M.; Wang, X.; Peng, X.; Feng, S.; Zhao, J.; Liao, L.; Zhang, Y.; Hou, Y.; Liu, J. Prognostic value of plasma von Willebrand factor levels in major adverse cardiovascular events: A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2020, 20, 72.
-
Kovacevic, K.D.; Mayer, F.J.; Jilma, B.; Buchtele, N.; Obermayer, G.; Binder, C.J.; Blann, A.D.; Minar, E.; Schillinger, M.; Hoke, M. Von Willebrand factor antigen levels predict major adverse cardiovascular events in patients with carotid stenosis of the ICARAS study. Atherosclerosis 2019, 290, 31–36.
-
Lv, J.-X.; Kong, Q.; Ma, X. Current advances in circulating inflammatory biomarkers in atherosclerosis and related cardio-cerebrovascular diseases. Chronic Dis. Transl. Med. 2017, 3, 207–212.
-
Van Belle, E.; Vincent, F.; Rauch, A.; Casari, C.; Jeanpierre, E.; Loobuyck, V.; Rosa, M.; Delhaye, C.; Spillemaeker, H.; Paris, C.; et al. von Willebrand Factor and Management of Heart Valve Disease. J. Am. Coll. Cardiol. 2019, 73, 1078–1088.
-
Springer, T.A. von Willebrand factor, Jedi knight of the bloodstream. Blood 2014, 124, 1412–1425.
-
Budde, U.; Pieconka, A.; Will, K.; Schneppenheim, R. Laboratory Testing for von Willebrand Disease: Contribution of Multimer Analysis to Diagnosis and Classification. Semin. Thromb. Hemost. 2006, 32, 514–521.
-
Stockschlaeder, M.; Schneppenheim, R.; Budde, U. Update on von Willebrand factor multimers: Focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul. Fibrinolysis 2014, 25, 206–216.
-
Zhang, Q.; Zhou, Y.-F.; Zhang, C.-Z.; Zhang, X.; Lu, C.; Springer, T.A. Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc. Natl. Acad. Sci. USA 2009, 106, 9226–9231.
-
Sadler, J.E. Biochemistry and Genetics of von willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424.
-
Denorme, F.; Vanhoorelbeke, K.; De Meyer, S.F. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Front. Immunol. 2019, 10, 2884.
-
Schneider, S.W.; Nuschele, S.; Wixforth, A.; Gorzelanny, C.; Alexander-Katz, A.; Netz, R.R.; Schneider, M.F. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. USA 2007, 104, 7899–7903.
-
Crawley, J.T.B.; de Groot, R.; Xiang, Y.; Luken, B.M.; Lane, D.A. Unraveling the scissile bond: How ADAMTS13 recognizes and cleaves von Willebrand factor. Blood 2011, 118, 3212–3221.
-
Sing, C.E.; Alexander-Katz, A. Elongational Flow Induces the Unfolding of von Willebrand Factor at Physiological Flow Rates. Biophys. J. 2010, 98, L35–L37.
-
Westein, E.; van der Meer, A.D.; Kuijpers, M.J.E.; Frimat, J.-P.; van den Berg, A.; Heemskerk, J.W.M. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 1357–1362.
-
Zheng, Y.; Chen, J.; López, J.A. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat. Commun. 2015, 6, 7858.
-
Zhang, C.; Kelkar, A.; Neelamegham, S. von Willebrand factor self-association is regulated by the shear-dependent unfolding of the A2 domain. Blood Adv. 2019, 3, 957–968.
-
Kanaji, S.; Fahs, S.A.; Shi, Q.; Haberichter, S.L.; Montgomery, R.R. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis: Hemostatic effect of platelet VWF in murine VWD. J. Thromb. Haemost. 2012, 10, 1646–1652.
-
Verhenne, S.; Denorme, F.; Libbrecht, S.; Vandenbulcke, A.; Pareyn, I.; Deckmyn, H.; Lambrecht, A.; Nieswandt, B.; Kleinschnitz, C.; Vanhoorelbeke, K.; et al. Platelet-derived VWF is not essential for normal thrombosis and hemostasis but fosters ischemic stroke injury in mice. Blood 2015, 126, 1715–1722.
-
Fujikawa, K.; Suzuki, H.; McMullen, B.; Chung, D. Purification of human von Willebrand factor–cleaving protease and its identification as a new member of the metalloproteinase family. Blood 2001, 98, 1662–1666.
-
Uemura, M.; Tatsumi, K.; Matsumoto, M.; Fujimoto, M.; Matsuyama, T.; Ishikawa, M.; Iwamoto, T.; Mori, T.; Wanaka, A.; Fukui, H.; et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood 2005, 106, 922–924.
-
Mannucci, P.M.; Capoferri, C.; Canciani, M.T. Plasma levels of von Willebrand factor regulate ADAMTS-13, its major cleaving protease. Br. J. Haematol. 2004, 126, 213–218.
-
Xiang, Y.; de Groot, R.; Crawley, J.T.B.; Lane, D.A. Mechanism of von Willebrand factor scissile bond cleavage by a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13). Proc. Natl. Acad. Sci. USA 2011, 108, 11602–11607.
-
Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 5620314.
-
Petri, B.; Broermann, A.; Li, H.; Khandoga, A.G.; Zarbock, A.; Krombach, F.; Goerge, T.; Schneider, S.W.; Jones, C.; Nieswandt, B.; et al. von Willebrand factor promotes leukocyte extravasation. Blood 2010, 116, 4712–4719.
-
Hillgruber, C.; Steingräber, A.K.; Pöppelmann, B.; Denis, C.V.; Ware, J.; Vestweber, D.; Nieswandt, B.; Schneider, S.W.; Goerge, T. Blocking von Willebrand Factor for Treatment of Cutaneous Inflammation. J. Investig. Dermatol. 2014, 134, 77–86.
-
Aymé, G.; Adam, F.; Legendre, P.; Bazaa, A.; Proulle, V.; Denis, C.V.; Christophe, O.D.; Lenting, P.J. A Novel Single-Domain Antibody against von Willebrand Factor A1 Domain Resolves Leukocyte Recruitment and Vascular Leakage during Inflammation—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1736–1740.
-
Khan, M.M.; Motto, D.G.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 reduces VWF-mediated acute inflammation following focal cerebral ischemia in mice: Role of ADAMTS13 and VWF in inflammatory brain injury. J. Thromb. Haemost. 2012, 10, 1665–1671.
-
De Meyer, S.F.; Savchenko, A.S.; Haas, M.S.; Schatzberg, D.; Carroll, M.C.; Schiviz, A.; Dietrich, B.; Rottensteiner, H.; Scheiflinger, F.; Wagner, D.D. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5217–5223.