Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Orphélie Lootens.
Mycotoxin contamination is a global food safety issue leading to major public health concerns. Repeated exposure to multiple mycotoxins not only has repercussions on human health but could theoretically also lead to interactions with other xenobiotic substances—such as drugs—in the body by altering their pharmacokinetics and/or pharmacodynamics. The combined effects of chronic drug use and mycotoxin exposure need to be well understood in order to draw valid conclusions and, in due course, to develop guidelines.
- food–drug interaction
- mycotoxins
- CYP450-enzymes
- human
- pharmacotherapeutics
- drugs
- PBPK-modelling
1. Introduction
Food contaminants comprise all unwanted dietary substances resulting from the applied cultivation conditions, production processes or environmental exposure. Certain contaminants pose a human health threat, such as toxic secondary metabolites produced by fungi, namely mycotoxins [1]. These toxins are detected in food crops such as maize, wheat, sorghum and peanuts [2]. They are produced by fungi as a self-protection mechanism during stressful conditions, and they are toxic to humans and animals, causing illness, and might even lead to death [3][4][5]. Fungi are able to produce multiple different mycotoxins, which lead to the existence of a great number of metabolites, exerting additive or even synergistic effects and causing (co)-morbidities and pathologies [5]. The exposure to these food contaminants is often chronic, depending on the geographical and climatic region of the world. High levels of contamination occur in regions where no strict regulations for mycotoxins are applied or where awareness is lacking, e.g., in low- and middle-income countries [6]. Research is imperative to study the impact of mycotoxin exposure on the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs taken concomitantly and vice versa. For the first time, a summary of the current knowledge on this topic is reported.
2. Mycotoxin–Drug Interactions Are an Emerging Research Topic
2.1. Food and Pharmacology: Food–Drug Interactions
Drugs interact with the diseased human body with the aim to bring benefit in a clinical context. However, a clinical condition, food, other drugs or a certain lifestyle may influence both the PK and PD of drugs. These influences may lead to a higher or lower effect of the drug, potentially leading to a changed clinical outcome. When drugs and food components are substrates for a certain enzyme, e.g., cytochrome P450 (CYP450) enzymes, a competitive or non-competitive interaction may occur when taken simultaneously. These interactions can be reversible or non-reversible, inhibitory or inducing [7]. The drug and food component may directly compete for metabolism via an enzyme, implying a competitive interaction. They may also bind to different sites of the enzyme, inducing a non-competitive interaction between the drug and food components through influencing a three-dimensional configuration of the enzyme’s active center. Food–drug interactions are an established area of research: nutrients can show a lower absorption due to interferences with concomitantly taken drugs. However, more importantly, at the level of metabolism and excretion there are interactions possible with potentially clinically relevant effects such as toxicity or ineffectiveness of the drug [8]. Drugs that interact with the metabolism of other exogenous compounds could also have a similar impact on the exposure as well as the effects of mycotoxins. Therefore, it is of utmost importance to thoroughly investigate the potential for mycotoxin–drug interactions, especially in a context where mycotoxin exposure is commonplace and highly prevalent, such as in low- and middle-income countries. Drugs of interest are, in this case, drugs that are commonly consumed in the areas where mycotoxin contamination is high (e.g., human immunodeficiency virus (HIV)-blockers in South Africa).2.2. Mycotoxin Metabolism through The CYP450 Complex
Mycotoxins—as with every xenobiotic—undergo absorption, distribution, metabolism and excretion in the human body. Metabolism occurs in different parts of the human body, but mainly by hepatic enzymes, and can be divided in phase I (oxidation, hydrolysis and reduction) [9], and phase II (conjugation) reactions [10]. Metabolism is a way to facilitate excretion from the body, among others, but it might lead to both detoxification and bioactivation of the parent compound. Considering all enzymes, the CYP450 complex is an important group involved in phase I metabolism, mainly found in the liver and gut. It is the most relevant enzyme family to consider, in view of its predominant role in drug metabolism, since it is involved in approximately 80 percent of all drug metabolism processes, as well as in the metabolism of endogenic compounds [11]. Variability in expression and activity of CYP450 enzymes is known to occur in humans and animals, mostly due to genetic polymorphisms [12]. In addition, factors such as age, gender, and health status have an impact on CYP450 enzyme expression and activity [13]. Different types of interactions are possible, potentially leading to higher or lower effects of the administered drugs or a less or more toxic effect of the mycotoxins one is exposed to. The most encountered mycotoxins and their metabolism are discussed below, i.e., AFs and Fusarium toxins.2.2.1. Aflatoxins
Metabolism of AFs via CYP450 results in different metabolites such as aflatoxin M1 (AFM1), aflatoxin Q1 (AFQ1), aflatoxin-exo-8,9-epoxide and aflatoxin-endo-8,9-epoxide (AFBO) [14]. AFQ1 and AFM1 are annotated as detoxified metabolites, whereas AFBO is considered a bioactivated metabolite, exerting carcinogenicity. CYP1A2 and CYP3A4 are the most important enzymes involved in the metabolism of AFB1 but also CYP3A5 and CYP3A7 play a role (Table 1) [14]. The intrinsic toxicity of AFB1 does not imply mutagenicity, though the epoxidation products of AFB1, AFBO, will form DNA-adducts and is therefore carcinogenic to humans. The DNA-adduct that will be formed is 8,9-dihydro-8-N7-guanyl-9-hydroxy aflatoxin (AFB1-N7-Gua), which is converted into AFB1-formamidopyrimidine (AFB1-FAPY), both leading to mutations [15]. Metabolism not only takes place in the liver and gut, but also in the respiratory tract after inhalation of, e.g., infected maize dust, where CYP2A13 will activate AFB1 to form the N7-guanine-adduct [16]. Large variations in AF metabolism have been observed both between and within species [17]. Age, gender and other factors are known to have an impact on the metabolism, making the extrapolation between species difficult [12].Table 1.
Summary of the main CYP-enzymes involved in the human metabolism of the listed mycotoxins.
| Mycotoxin | Involved CYP-Enzymes | References |
|---|---|---|
| AFB1 | CYP3A4, CYP3A5, CYP3A7, CYP1A2, CYP2A13 | [14][15][16][17] |
| DON | No phase I metabolism | [18][19][20][21][22] |
| T-2 | CYP3A4, CYP2E1, CYP1A2, CYP2B6, CYP2D6 and CYP2C19 | [23][24] |
| ZEN | CYP3A4, CYP3A5, CYP2C8, CYP1A2 | [25][26] |
| FB1 | No phase I metabolism | [27][28][29][30] |
| ENN B1 | CYP3A4, CYP3A5, CYP1A2, CYP2C19 | [31][32] |
2.2.2. Fusarium Toxins
Via phase II metabolism, DON is converted to DON-3-sulfate in the intestine, liver and kidney in poultry, and is rapidly eliminated from the body via excreta [18]. Animals produce more DON-metabolites compared to humans; only pigs show similar DON toxicokinetics and metabolism [19]. In the human body, a part of the consumed DON-fraction is metabolized to DON-glucuronides in the liver and gut by phase II uridine diphosphate-glucuronosyltransferase enzymes (UGT) [20]. More than 75 percent of the DON found in urine appeared to be in a glucuronidated form. Warth et al. (2013) [21] and Sayyari et al. (2018) [22] concluded that CYP3A4 was not involved in the metabolism of DON [21][22]. Moreover, none of the CYP450 enzymes are involved in the metabolism of DON [17]. DON-15-glucuronide (DON-15-Glc) is the most abundant DON metabolite found in human urine [20]. Other DON-metabolites such as iso-DON-8-glucuronide (iso-DON-8-Glc), iso-DON-3-glucuronide (iso-DON-3-Glc) and de-epoxy deoxynivalenol-3-glucuronide (DOM-3-Glc) have been identified in urine of humans. Those metabolites were also found in pig urine and plasma, but in very low concentrations, whereas DON-3-Glc and DON-15-Glc appear to be the dominating metabolites [20][21][22]. With the same molecular backbone as DON, T-2 belongs to the trichothecene family. T-2 gets rapidly metabolized into a number of metabolites, mainly phase I metabolites, e.g., 3′-OH-T-2, 3′-OH-HT-2, HT-2, T-2 triol and glucuronides thereof (phase II) [17]. Another study that focused on ruminants and non-ruminants showed that the initial step in T-2 biotransformation is deacetylation leading to the formation of HT-2 [23]. Carboxylesterases are the most important enzymes for the metabolism of T-2, followed by CYP3A4, CYP2E1, CYP1A2, CYP2B6, CYP2D6 and CYP2C19 of the CYP450 complex, as shown in in vitro experiments in human liver microsomes [24]. Via phase I metabolism ZEN is converted to α-zearalenol (α-ZEL) and β-zearalenol (β-ZEL) via the enzymes α-hydroxysteroid dehydrogenase and β-hydroxysteroid dehydrogenase, respectively, as well as via CYP-enzymes, and phase II metabolism to glucuronides thereof. The intestine is the most important site of ZEN metabolism after oral ingestion. In vitro tests with human CYP450 enzymes demonstrated the oxidation of ZEN via CYP2C8, CYP3A4 and CYP3A5. An inhibition of CYP2C and CYP3A was observed due to the presence of ZEN [25]. Another study discovered that CYP3A4 and CYP1A2 are responsible for the hydroxylation of ZEN into catechol metabolites. This study proved that aromatic hydroxylation of ZEN is the main metabolic pathway in vitro [26]. Unlike AFB1, FBs do not get extensively metabolized. In rats, the majority of consumed FBs were excreted in their unchanged form. A study has also been performed in monkeys where hydrolysis products of FB1 have been observed in the feces [27]. FB1 causes a small increase in CYP1A activity and expression [28]. Another study showed that there is no phase I metabolism of FB1, but that the intestinal microbiota in pigs and poultry can hydrolyze FB1 [29][30]. Using two in vitro systems, namely human liver microsomes (HLM) and CYP3A4-containing nanodiscs (ND), it was demonstrated that ENN B1 is mainly metabolized by CYP3A4 and CYP3A5 [31]. Thise study identified 11 metabolites of ENN B1. In addition, DON was studied, clarifying that it was not metabolized by CYP3A4, though it did interact with the metabolism of ENN B1, since DON decreased its metabolic rate. This finding indicated that DON may cause a non-competitive inhibition of CYP3A4, leading to a slower metabolism of ENN B1. A comparative in vitro and in vivo metabolism study of ENN B1 in pigs showed that it was extensively metabolized in pig liver microsomes, which was confirmed in vivo after both intravenous and oral administration. The main metabolites were observed in higher levels after oral administration as compared to intravenous administration. This indicates that pre-systemic metabolism contributed to ENN B1′s metabolism when taken per os [31]. A different study was performed, where the metabolism of ENN B1 was confirmed in microsomes of rats, dogs and humans. Additionally, phenotyping of the CYP450 enzymes was performed using chemical inhibitors, which are selective for specific human enzymes. CYP3A4 was the main enzyme involved in the metabolism, followed by CYP1A2 and CYP2C19. This confirmed the results of the previously discussed studies where CYP3A4 was pinpointed to be the principal enzyme involved in the metabolism of ENN B1 [32]. As shown in Table 1, the hepatic biotransformation of certain mycotoxins is well understood. It is clear that chronic exposure to mycotoxins may potentially lead to an altered biotransformation of xenobiotics or vice versa, such as drugs, in the liver and/or the gut through interaction at the level of the biotransformation enzymes. Certain mycotoxins are metabolized via CYP450 enzymes; all drugs that are known CYP450 perpetrators and/or substrates will have an impact on the metabolization of mycotoxins, wherein the same CYP450 enzymes are involved. The formation of less (inhibition) or more (induction) metabolites is crucial for mycotoxins and the exertion of their toxicity, taking into account the biochemical activation of some mycotoxins (e.g., AFB1 is metabolized into the more toxic AFBO via CYP450 enzymes). Figure 1 pinpoints the different pathways of ENN B1, T-2, ZEN and AFB1, as a CYP450 substrate or CYP450 perpetrator, to their metabolites. Drugs that are strong CYP3A4-inducers (e.g., carbamazepine, an anti-epileptic drug) or strong CYP3A4-inhibitors (e.g., lopinavir, a human immunodeficiency blocker) will potentially impact the metabolism of the mentioned CYP3A4-substrate mycotoxins. Inducers will lead to more metabolite formation, which is satisfactory in case of detoxification, but not in case of bioactivation (e.g., AFBO formation). Inhibitors will lead to less metabolite formation.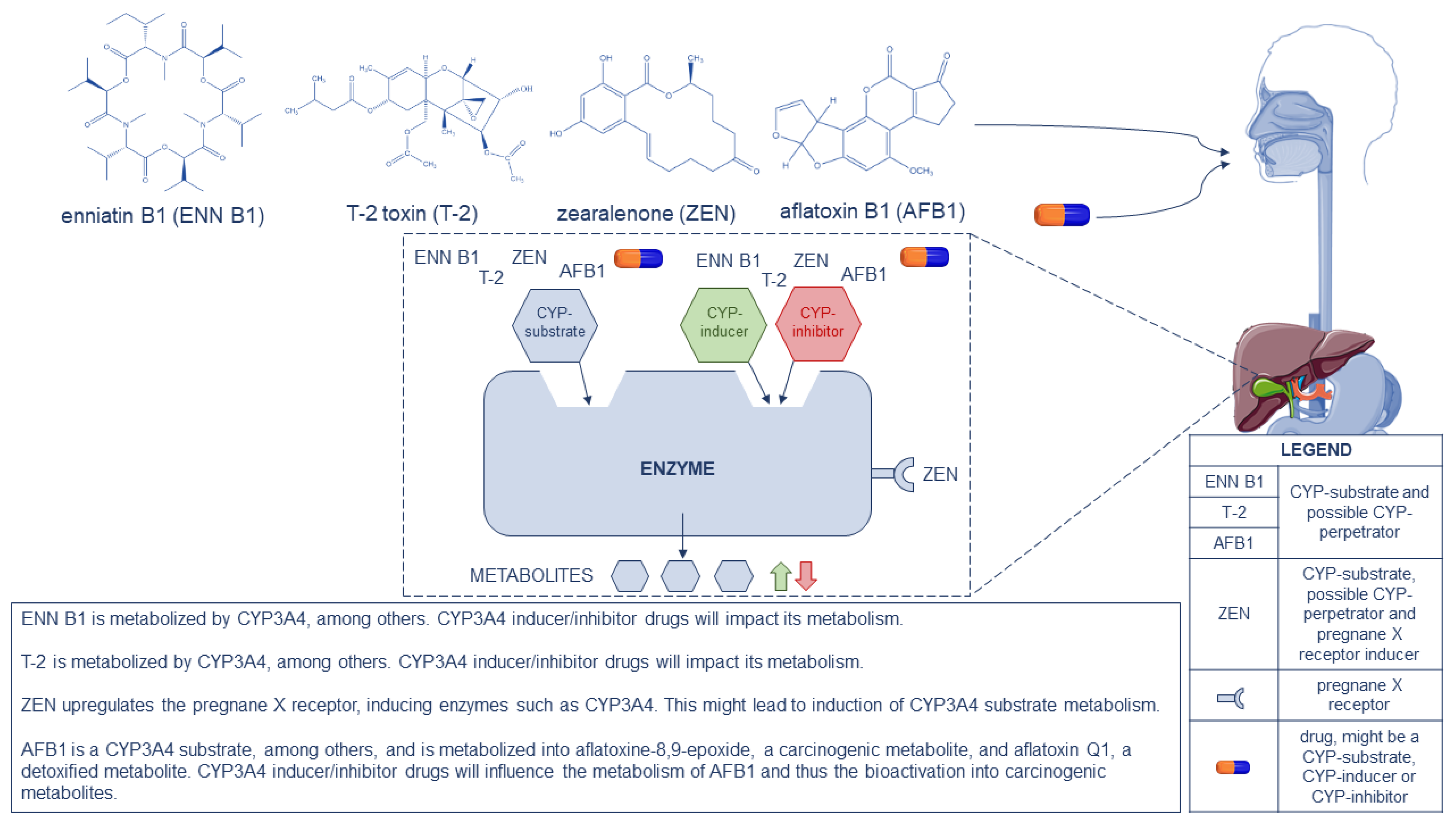
Figure 1. Representation of enniatin B1 (ENN B1), T-2 toxin (T-2), zearalenone (ZEN), aflatoxin B1 (AFB1) and drugs as CYP450 substrates and potential CYP450 perpetrators, following the gastrointestinal tract to the liver where metabolism occurs. A schematic drawing is given on the possible pathways that might take place in the human liver. In the bottom-right corner, a legend is displayed. At the bottom-center an explanation is given for ENN B1, T-2, ZEN and AFB1, as CYP3A4 substrates and as pregnane X receptor inducer in case of ZEN, on the possible impact on metabolism.
References
- Smith, M.C.; Madec, S.; Coton, E.; Hymery, N. Natural Co-occurrence of mycotoxins in foods and feeds and their in vitro combined toxicological effects. Toxins 2016, 8, 94.
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ‘FAO estimate’ of 25%. Crit. Rev. Food Sci. Nutr. 2019, 60, 2773–2789.
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516.
- Pleadin, J.; Frece, J.; Markov, K. Mycotoxins in food and feed. Adv. Food Nutr. Res. 2019, 89, 297–345.
- Sobral, M.; Faria, M.; Cunha, S.; Ferreire, I. Toxicological Interactions between Mycotoxins from Ubiquitous Fungi: Impact on Hepatic and Intestinal Human Epithelial Cells. Chemosphere 2018, 202, 538–548.
- Kebede, H.; Liu, X.; Jin, J.; Xing, F. Current status of major mycotoxins contamination in food and feed in Africa. Food Control 2020, 110, 106975.
- Bushra, R.; Aslam, N.; Khan, A.Y. Food-drug interactions. Oman Med. J. 2011, 26, 77–83.
- Weininger, J. Nutritional Disease—Food-Drug Interactions|Britannica. 2016. Available online: https://www.britannica.com/science/nutritional-disease/Foodborne-illnesses (accessed on 6 December 2019).
- Zhou, S.-F.; Liu, J.-P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295.
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. 2010, 154, 103–116.
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 systems—Biological variations of electron transport chains. Biochim. Biophys. Acta—Gen. Subj. 2007, 1770, 330–344.
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.B.; Tien, Y.C.; Jeong, H.; Pan, X.; Shireman, L.M.; et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab. Dispos. 2016, 44, 343–351.
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036.
- Deng, J.; Zhao, L.; Zhang, N.Y.; Karrow, N.A.; Krumm, C.S.; Qi, D.S.; Sun, L.H. Aflatoxin B1 metabolism: Regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat. Res. Rev. Mutat. Res. 2018, 778, 79–89.
- Bbosa, G.S.; Kitya, D.; Odda, J.; Ogwal-Okeng, J. Aflatoxins metabolism, effects on epigenetic mechanisms and their role in carcinogenesis. Health 2013, 5, 14–34.
- Jolly, P.; Jiang, Y.; Ellis, W.; Awuah, R.; Nnedu, O.; Phillips, T.; Wang, J.-S.; Afriyie-Gyawu, E.; Tang, L.; Person, S.; et al. Determinants of aflatoxin levels in Ghanaians: Sociodemographic factors, knowledge of aflatoxin and food handling and consumption practices. Int. J. Hyg. Environ.-Health 2006, 209, 345–358.
- Vidal, A.; Mengelers, M.; Yang, S.; De Saeger, S.; De Boevre, M. Mycotoxin Biomarkers of Exposure: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1127–1155.
- Schwartz-Zimmermann, H.E.; Fruhmann, P.; Dänicke, S.; Wiesenberger, G.; Caha, S.; Weber, J.; Berthiller, F. Metabolism of deoxynivalenol and deepoxy-deoxynivalenol in broiler chickens, pullets, roosters and turkeys. Toxins 2015, 7, 4706–4729.
- Schelstraete, W. Interactions between Fusarium Mycotoxins and Cytochrome P450 Drug Metabolizing Enzymes in a Porcine Animal Model. Available online: https://biblio.ugent.be/publication/8632658/file/8632659 (accessed on 22 August 2022).
- Vidal, A.; Claeys, L.; Mengelers, M.; Vanhoorne, V.; Vervaet, C.; Huybrechts, B.; De Saeger, S.; De Boevre, M. Humans significantly metabolize and excrete the mycotoxin deoxynivalenol and its modified form deoxynivalenol-3-glucoside within 24 hours. Sci. Rep. 2018, 8, 5255.
- Warth, B.; Sulyok, M.; Berthiller, F.; Schuhmacher, R.; Krska, R. New insights into the human metabolism of the Fusarium mycotoxins deoxynivalenol and zearalenone. Toxicol. Lett. 2013, 220, 88–94.
- Sayyari, A.; Kruse Faeste, C.; Hansen, U.; Uhlig, S.; Framstad, T.; Schatzmayr, D.; Sivertsen, T. Effects and biotransformation of the mycotoxin deoxynivalenol in growing pigs fed naturally-contaminated grain pelleted with and without the addition of Coriobacteriaceum DSM 11798. Food Addit. Contam. Part A 2018, 35, 1394–1409.
- Kuca, K.; Dohnal, V.; Jezkova, A.; Jun, D. Metabolic Pathways of T-2 Toxin. Curr. Drug Metab. 2008, 9, 77–82.
- Lin, N.N.; Chen, J.; Xu, B.; Wei, X.; Guo, L.; Xie, J.W. The roles of carboxylesterase and CYP isozymes on the in vitro metabolism of T-2 toxin. Mil. Med. Res. 2015, 2, 13.
- Bravin, F.; Duca, R.C.; Balaguer, P.; Delaforge, M. In Vitro cytochrome P450 formation of a mono-hydroxylated metabolite of zearalenone exhibiting estrogenic activities: Possible occurrence of this metabolite in Vivo. Int. J. Mol. Sci. 2009, 10, 1824–1837.
- Pfeiffer, E.; Hildebrand, A.; Damm, G.; Rapp, A.; Cramer, B.; Humpf, H.-U.; Metzler, M. Aromatic hydroxylation is a major metabolic pathway of the mycotoxin zearalenone in vitro. Mol. Nutr. Food Res. 2009, 53, 1123–1133.
- Shoshei, S.; Makoto, K. Fumonisin B1—An Overview|ScienceDirect Topics. 2010. Available online: https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/fumonisin-b1 (accessed on 9 December 2019).
- Mary, V. Effects of AFB1, FB and their mixture on the aryl hydrocarbon receptor and CYP450 1A induction. Food Chem. Toxicol. 2015, 75, 104–111.
- Fodor, J.; Meyer, K.; Gottschalk, C.; Mamet, R.; Kametler, L.; Bauer, J.M.; Horn, P.; Kovacs, F.; Kovacs, M. In vitro microbial metabolism of fumonisin B1. Food Addit. Contam. 2007, 24, 416–420.
- Antonissen, G.; De Baere, S.; Novak, B.; Schatzmayr, D.; den Hollander, D.; Devreese, M.; Croubels, S. Toxicokinetics of Hydrolyzed Fumonisin B1 after Single Oral or Intravenous Bolus to Broiler Chickens Fed a Control or a Fumonisins-Contaminated Diet. Toxins 2020, 12, 413.
- Ivanova, L.; Uhlig, S.; Devreese, M.; Croubels, S.; Fæste, C.K. Biotransformation of the mycotoxin enniatin B1 in pigs: A comparative in vitro and in vivo approach. Food Chem. Toxicol. 2017, 105, 506–517.
- Fæste, C.K.; Ivanova, L.; Uhlig, S. In vitro metabolism of the mycotoxin enniatin B in different species and cytochrome P450 enzyme phenotyping by chemical inhibitors. Drug Metab. Dispos. 2011, 39, 1768–1776.
More