Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Michael Fossel and Version 2 by Dean Liu.
Interventions aimed at the cellular origin of the pathology would have a better chance of preventing and treating cardiovascular disease, lowering healthcare costs, and incurring less risk.
- cell senescence
- aging
- cardiovascular disease
- telomerase
- telomere
1. The Role of Cell Aging in Age-Related Cardiovascular Disease
Cell aging has been theorized [1][2][3][4][53,54,56,60] and implicated in age-related disease in animals [5][6][61,62] and humans [7][8][50,63]. The unified systems model (Figure 12) suggests that cell aging mediates the cascade of events between upstream clinical risk factors and downstream clinical outcomes. Traumatic injury may accelerate cell aging in joints, UV exposure may accelerate cell aging in the skin, and having two APOE4 alleles may accelerate microaggregate formation, even in the context of relatively mild microglial cell aging, resulting in earlier and more severe Alzheimer’s disease. The downstream heterogeneity of clinical outcomes (specific diagnosis, age of onset, course of disease, individual clinical findings, etc.) results from the upstream heterogeneity of genetic, epigenetic, and behavioral history. However, pathology plays out in the unified “stage” of cell aging (Figure 12).
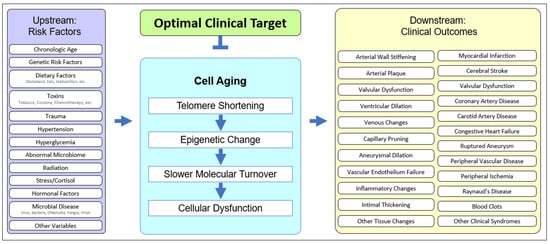
Figure 12. The unified model of cell aging being central to age-related cardiovascular disease. Upstream risk factors provide variable starting points, affect cell aging, and result in variable downstream clinical outcomes, suggesting an optimal point of intervention.
Cell aging is typified by a deceleration in molecular turnover, whether intracellular, extracellular, or intranuclear, and whether protein, lipid, or other molecular moieties, resulting in a gradual increase in the percentage of dysfunctional molecules. Slower molecular turnover results in an increased proportion of damage, but the rate of damage can also increase as a result of genetic or environmental effects acting synergistically with cell aging. Upstream risk factors can not only increase the rate of cell aging [9][10][11][12][64,65,66,67] but can also exacerbate the effects of cell aging. For example, rheologic trauma (e.g., at bifurcations, the aortic arch, etc.) and barotrauma (e.g., hypertension) may increase the rate of cell aging and thereby hasten the onset of pathology (e.g., aortic aneurysm), but genetic risks are also operative [13][14][15][16][68,69,70,71].
Cardiovascular disease is the result of vascular endothelial cell aging [17][18][72,73], as well as aging in smooth muscle cells [19][20][74,75], cardiomyocytes [21][76], cardiac fibroblasts [22][77], and immune cells [23][78]. In aging endothelial cells, there is a decreased endothelial nitric oxide synthase (eNOS) and nitric oxide, critical in atherogenesis and hypertension. Endothelial cell aging increases monocyte adhesion, implicated in atherogenesis, and the induction of endothelial cell aging results in typical atherogenic changes. Importantly, these processes are linked with telomerase activity. In the case of laminar fluid stress, telomerase is required for normal endothelial cell response [24][79], while telomere shortening is associated with increasing arterial stiffness [25][80]. This vascular dysfunction is ameliorated or normalized by telomerase, causing reversion to a pattern of gene expression typical of younger endothelial cells [26][27][81,82], indicting endothelial cell aging in atherogenesis [28][83]. Upstream risk factors leading to cell aging and downstream outcomes predisposing to cardiovascular disease are important to integrate into the Unified Model.
2. Upstream Risk Factors in Cardiovascular Disease
Upstream risk factors are clinical starting points that operate within the context of cell aging and cause age-related disease. Pathology can result from normal cell aging, from accelerated cell aging (e.g., hypertension, diabetes, vascular infections, tobacco use, poor diet, etc.), or from accelerated molecular damage (e.g., genetically abnormal free radical scavengers, membrane lipids, mitochondrial enzymes, etc.). Upstream risks may be categorized as genetic, epigenetic, or behavioral.
Genetic risks may be relatively asymptomatic in early life, as in patients with aberrant elastin or collagen genes [29][84], but increasingly symptomatic in later life as molecular pools are recycled far more slowly and the percentage of damaged molecules rises. Risks may be genetic, as with abnormal cytochrome C oxidase or mitochondrial genes [30][31][85,86], but interactive processes may also occur, for example in endothelial cell aging [12][32][33][67,87,88] and aortic smooth muscle cells [34][89], where oxidative damage [11][66] and cell aging work in tandem [35][90]. Epigenetic risks may include regulatory elements (which greatly outnumber protein-expressing genes), including variability in inherited telomere lengths [36][91]. Epigenetic differences manifest as regulatory idiosyncrasies: patients with identical genes may have different patterns of expression and resulting differences in age-related disease.
Telomere length varies among newborns, while it is similar in different organs of the human fetus [37][92]. Genetic mechanisms involved in the regulation of TERT expression and telomerase activity are described in recent reviews [38][93]. Germline mutations in TERT and other genes that control chromosome termini result in the shortening of telomeres transmitted to the progeny. This leads to progressive telomere length changes over successive generations. Somatic mutations in telomerase machinery, as well as the epigenetic downregulation of TERT and its co-activators, account for tissue-specific differences in telomere attrition and senescence onset during aging. Signaling pathways including WNT, converging on KLF4, as well as c-MYC, GA-binding proteins (GABP), and other E26 transformation-specific (ETS) family transcription factors have been found to regulate TERT transcription [39][94]. Epigenetic mechanisms also include promoter methylation, histone deacetylase inhibitors, and miRNAs [40][95]. The post-translational regulation of TERT activity is also important to consider [41][96]. The age of telomerase inactivation predetermines the rate of telomere attrition, which varies among individuals [36][91]. In combination with somatic genetic and epigenetic changes in the oxidative damage protection pathways, these variables in turn predetermine senescence onset in distinct cell populations and the resulting aging of individual tissues.
Behavioral risks (e.g., smoking [42][43][44][97,98,99], air pollution [45][46][47][48][11,100,101,102], diet [49][103], alcohol, etc.) may either increase the rate of cell aging or exacerbate the effects of cell aging. An example from the central nervous system may clarify. Head trauma or CNS infections would increase the risk of Alzheimer’s disease by increasing the rate of cell aging, while a patient with no history of trauma or infection but who is biallelic for APOE4 is prone to earlier microaggregate formation, exacerbating the effects of cell aging. There is a growing body of evidence that the rate of telomere shortening is also a function of environment and behavior.
Cell aging is universal, while the risk, timing, and severity of the resulting age-related cardiovascular diseases are defined by genetic, epigenetic, and behavioral starting points. Specific downstream clinical outcomes—myocardial infarction, stroke, varicosities, cardiomyopathy, etc.—depend on the specific upstream starting points.
3. Downstream Outcomes in Cardiovascular Disease
Downstream outcomes—biomarkers, clinical syndromes, or pathology—vary because upstream risk factors vary, even in the face of a unified fundamental process, that of cell aging. In atherosclerosis, for example, the initial formation of a fatty streak, composed of lipid-laden macrophages (foam cells) within the intima, is followed by vascular smooth muscle cell proliferation, fibrous plaques, cholesterol deposits, and inflammatory cell recruitment [50][104]. The observable fatty streak formation is, however, preceded by a less obvious inflammatory response within the endothelium, which has been linked to a number of insults, including hypertension, diabetes, tobacco use, and the inflammatory secretory pattern directly associated with cell aging [51][105]. This results in the recruitment of inflammatory cells (e.g., monocytes) and the loss of adhesion in endothelial cell junctions. The failure of cell junction integrity causes the leakage of plasma components, including LDL cholesterol, into the subendothelial compartment. Inflammation further contributes to endothelial damage and predisposes to LDL phagocytosis. Recruited monocytes transform into macrophages, phagocytosing oxidized LDL and forming atherosclerotic foam cells [50][104]. They also secrete inflammatory factors, induce smooth muscle cell proliferation and often form fibrotic tissue.
The earliest fatty streaks in children and adolescents lack this fibrous component and other pathological features that characterize atherosclerosis in elderly patients where cell aging is prominent [51][52][53][105,106,107]. Pathology can occur without cell senescence, for example in the presence of significant physical or oxidative damage to the endothelium, as evidenced by arterial fatty streaks in many patients by age twenty. However, the arterial remodeling typical of the young [51][52][53][54][105,106,107,108] is slower or absent as cell aging progresses and other changes, such as mitochondrial dysfunction, occur [21][55][56][76,109,110]. Cell aging drives the progression of atherosclerosis even without classical risk factors [57][58][111,112] and is critical to the progression of significant pathology [53][54][107,108]. Early lesions may regress with effective clinical intervention in upstream risk factors, but advanced lesions, in which cell aging is prominent, are not [52][57][58][59][106,111,112,113]. Even aggressive intervention in risk factors (e.g., diabetes, hypertension, and cholesterol) are insufficient, as the cell aging of the arterial cells previously induced by such factors continues apace [23][28][50][52][57][60][61][78,83,104,106,111,114,115]. The vascular endothelial cell aging continues, and pathology accelerates [23][28][57][60][61][78,83,111,114,115].
Hutchinson-Gilford progeria provides an example of the role of cell aging and telomere shortening in atherosclerosis. In these children, their lamin-A mutation results in telomere lengths typical of 70-year-old patients. Although lacking classical risk factors—diabetes, tobacco use, hypertension, or elevated cholesterol—they develop atherosclerotic pathology [62][116] and usually die (average lifespan 12.7 years) of atherosclerotic disease [63][64][117,118]. Mouse models of H g progeria likewise show an impairment of mechano-signaling in endothelial cells, suggesting that cell aging and their resulting senescence contributes to excessive fibrosis and cardiovascular disease [65][119], and studies on human progeric cells suggest that endothelial-targeted therapy may be effective [66][120]. Cardiovascular disease occurs in the context of accelerated cell aging, even when other risk factors are absent.
Cardiovascular risk factors such as diabetes, hypertension, obesity, and dyslipidemia may be promoted by cell aging and senescence, but may also accelerate cell aging, resulting in a vicious cycle of pathology. Ongoing damage accelerates cell aging, leading to subendothelial exposure, persistent and irreversible inflammation, the progression of atherosclerotic plaque, reduced fibrous plaque coverage, and eventual rupture [23][28][60][61][78,83,114,115]. This is all the more serious with LDL elevation, as high cholesterol leads to increased plaque and the oxidation of LDL within the intima, with increased inflammatory responses [52][57][58][59][106,111,112,113].
A similar causal cascade occurs in aneurysms, as the cell aging of the vascular walls results in structural deterioration. The vascular mesenchymal stromal cells (VMSCs) from aortic aneurysms are more senescent than those from healthy aortas, and medial smooth muscle cells taken from aneurysms are likewise more senescent than cells from normal arteries in the same individual [67][68][69][121,122,123].
Cardiac failure is also linked to cell aging. Cardiomyocytes [70][71][48,124] and cardiac fibroblasts [22][77] display the hallmarks of cell aging, including dysfunctional DNA repair, mitochondrial dysfunction, impaired contractility, and increased fibrosis, with the subsequent impairment of ejection fraction and heart failure [21][71][72][73][76,124,125,126]. Curiously, experimentally induced transient [74][127] or premature senescence [75][128] may reduce perivascular fibrosis and inflammation, perhaps by converting actively dysfunctional (though aged) cells into far less active, replicative senescent cells. Age-related increases in arrhythmias may also be linked to cell aging [76][129].
4. Heterogeneity of Clinical Presentations in Cardiovascular Disease
Age-related cardiovascular disease is clinically heterogenous, encompassing a spectrum of diseases that share a temporal predilection (aging) and a tissue of origin (the cardiovascular system) but often little else, at least superficially. There may appear to be little in common between strokes, myocardial infarctions, varicose veins, and capillary pruning. However, such diseases all demonstrate vascular endothelial cell aging and secondary organ pathology [77][130].
In the case of CNS arterial disease, both hemorrhagic and thrombotic stroke can result from the cell aging of the vascular endothelial cells, the former from arterial rupture and the latter from arterial blockage. In the case of coronary disease, myocardial infarction results from insufficient flow secondary to vascular endothelial cell aging. Other presentations typical of aging patients [78][131]—varicose veins, chronic venous insufficiency, deep vein thrombosis, capillary pruning, etc.—can be the result of vascular endothelial cell aging, with the consequent loss of integrity and structural resilience, whether in arterial, venous, or capillary walls.
Clinical heterogeneity is common. Patients of identical age may vary in disease, vascular localization, onset, or course, and this heterogeneity likely results from the heterogeneity of the initial risk factors. Individuals differ genetically, epigenetically, and behaviorally. Genetic differences may include alleles which impact cholesterol metabolism, inflammatory processes, immune responses, the efficacy of hepatic detoxification, renal physiology, collagen strength, elastin production, or a myriad of other genetic “starting points” prior to the changes incurred by cell aging. Epigenetic differences may be equally profound: rather than an abnormal gene, there is an abnormal pattern of gene expression, with subtly different responses to physiologic stimuli. Behavioral history—alcohol use, diet, exercise, infectious disease, trauma, etc.—also generates clinical heterogeneity in cardiovascular disease.
Genetic risk factors have become increasingly well-established over the past decade with the increased use of genome-wide association studies (GWAS) to identify disease-susceptibility loci [79][80][81][132,133,134]. However, epigenetic diagnostic markers and therapeutic targets, independent of genetic inheritance, have also been identified [82][83][84][135,136,137]. While associations between the risk factors and the clinical outcomes are commonly acknowledged, the cascade of events proceeding via cell aging, presenting as overt changes in tissue and organ function, and ultimately as clinical disease, are less well appreciated.
Contributing to the confusion, many studies analyze aging in cells that are not responsible for the disease [85][86][138,139]. Because of sample collection simplicity, circulating leukocytes rather than vascular endothelial cells are commonly analyzed for telomere attrition [87][88][89][90][91][92][93][94][95][96][140,141,142,143,144,145,146,147,148,149]. As might be expected, there is a rough correlation in telomere lengths between different tissues since the entire organism is undergoing progressive cell aging over time. However, the same might be said of the correlation between cardiovascular disease, CNS neurodegenerative disease, osteoarthritis, osteoporosis, and other age-related diseases as the patient ages. The correlation reflects the age of the patient but is not sufficiently specific to the tissue in question. Although too often measured in patients with cholesterol abnormalities [97][150] or cardiovascular disease [98][99][100][101][102][103][151,152,153,154,155,156], leukocyte telomere lengths can change for independent reasons, such as infections or autoimmune reactions. Relying upon leukocyte measures in assessing cardiovascular disease results in unwarranted conclusions based on inappropriate correlations and irrelevant cell choices. In assessing risk factors for age-related cardiovascular disease, such as diabetes [104][105][157,158] or vascular aging [106][159] rwesearchers se see the same error in measuring telomeres in leukocytes [107][108][160,161] rather than in relevant cells. Because vascular dysfunction in cardiovascular disease is largely attributable to endothelial cells [35][90] and/or vascular smooth muscle cells [20][75], telomere lengths in such cells should be used as a gauge of cell aging and atherosclerosis [109][162]. Studies that focus on the measurement of telomeres and cell aging of the appropriate cells find a clearer picture [110][111][112][163,164,165]. The data show (and reviews have emphasized) that telomere shortening and cell aging are not only implicated in age-related cardiovascular aging but precede and are more prominent in affected tissues [113][114][166,167]. These studies underline the role of cell aging in multiple age-related cardiovascular diseases [115][168], including hypertension [116][117][169,170], and atherosclerosis [28][118][119][83,171,172], predisposing to heart failure.
Identifying cell aging as a fundamental mechanism underlying cardiovascular disease offers a more complex—but perhaps a more accurate—paradigm of disease etiology. Current diagnostic approaches to cardiovascular disease test for either upstream risk factors or downstream biomarkers. High cholesterol [97][120][150,173] or hypertension, for example, are clearly upstream risk factors but may operate through the acceleration of cell aging [35][121][122][123][90,174,175,176]. Coronary calcium scans [124][177], on the other hand, are a downstream biomarker, but cell aging may be the driving force behind plaque buildup [125][178].
Upstream risk factors such as chronological age, genetic risk, diet, stress, and trauma contribute to cell aging, while downstream biomarkers of cardiovascular disease, such as venous changes, arterial wall hardening, and myocardial infarctions are the clinical consequences of cell aging. Changes in endothelial physiology, including the eNOS/NO and FGF21 pathways, as well as calcium signaling, appear to be detrimental [32][87]. Other metabolic changes, including hypertensive or hyperglycemic injury to these cells [120][173], are also important [126][179]. Cardiovascular cell aging can also cause secondary CNS disease [127][180], whether as an acute insult (e.g., stroke) or as a chronic result of vascular insufficiency and changes in the blood–brain barrier. Indeed, telomere shortening and cell aging are prominent not only in Alzheimer’s but also in vascular dementia and other age-related neurodegenerative diseases [128][129][130][181,182,183].
There is accumulating evidence that vascular endothelium aging underlies age-related cardiovascular pathology [8][125][63,178]. Endothelial aging induced by experimental telomere attrition stimulates metabolic disorders through SASP [56][131][110,184]. Healthy angiogenesis is not an active process in most mature organs, yet endothelial cell division and capillary sprouting underlie normal maintenance in muscle, adipose tissue, and likely the brain. Muscle regeneration relies on angiogenesis, and dysfunctional angiogenesis is implicated in some cardiovascular diseases. Endothelial cell aging is also likely to underlie peripheral artery disease (PAD) and capillary pruning. Although cause-and-effect may be complicated [132][133][134][135][185,186,187,188], cell aging offers a novel interventional target in both PAD and type-2 diabetes (T2D) [136][137][189,190]. While most cardiac diseases may be attributable to vascular endothelial cell aging, other cell types are also implicated. Cardiomyopathy may be the result of cardiac fibroblast aging [138][191]. Pericytes, mural cells [139][192], macrophages [140][193], and other myeloid cells (with foam cell and plaque formation) may also be involved [141][194]. Mice lacking TERT in perivascular progenitor cells [142][195] have premature metabolic disease [143][196], more so if given a high-caloric diet [144][197].
Adipose tissue, a key reservoir of senescent cells, may indirectly contribute to the aging of cardiovascular cells [145][146][147][148][198,199,200,201]. In obesity, the death of hypertrophic and hypoxic adipocytes prompts leukocyte infiltration, inflammation, fibrosis, and reduced ability to contain triglycerides, which results in steatosis and may contribute to the development of T2D [149][202]. Early in life, adipocyte progenitor cells divide continuously [150][151][203,204], replacing dysfunctional hypertrophic adipocytes [152][153][154][205,206,207]. With age, adipocyte progenitors lose replicative capacity, which is accelerated by hyperlipidemia [97][155][156][150,208,209], obesity [157][158][159][160][210,211,212,213], and oxidative stress [161][162][214,215]. Telomere length in progenitor cells defines the ability of adipose tissue to expand by hyperplasia, rather than hypertrophy [144][197], perhaps explaining the lack of T2D in “healthy obese” individuals [143][196]. In mice lacking TERT, adipocyte progenitors undergo premature replicative senescence, and T2D development is accelerated [142][195]. Obesity development also involves increased endothelial cell division (as well as leukocyte infiltration) to vascularize expanding adipose tissue [143][196]. The accelerated cell aging may aggravate adipose dysfunction, the accompanying dyslipidemia [163][216], and hypertension [164][217], independently of the cardiovascular risk factors [165][218]. All of these aging cells secrete SASP factors with systemic vascular effects. Aging monocytes from adipose tissue may also disseminate systemically and may contribute to atherosclerosis and inflammation directly. Potential long-distance effects of other types of aging cells in adipose tissue remain to be investigated [166][219].