Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Elena Kaznacheyeva and Version 2 by Conner Chen.
Quinazoline derivatives are a large pool of natural and synthetic compounds. The first derivatives of quinazoline were synthesized at the end of the 19th century. one quinazoline derivative (4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine)—EVP4593 (also marked as QNZ) was originally synthesized in 2003 as a modulator of the nuclear factor kappa B (NF-κB) signal transduction pathway. Since that time, EVP4593 has been widely used as a blocker of NF-κB signaling (Sigma-Aldrich, cat #481417). Further it has been reported the ability of EVP4593 to affect store-operated calcium channels. Here we discussed molecular mechanisms underlying the EVP4593 functioning in the cells.
- quinazoline derivatives
- QNZ
- EVP4593
- calcium signaling
- NF-κB signaling
1. EVP4593 and NF-κB Signaling
The nuclear factor kappa B (NF-κB) signaling pathway is one of the general signal transduction pathways that mediate gene expression of proinflammatory and antiapoptotic factors, cell proliferation, differentiation, adhesion, migration and angiogenesis [1][14]. The hyperactivation of NF-κB signaling is a hallmark of many types of cancer [1][14] and inflammatory-associated pathologies [1][2][14,15]. Additionally, the chronic inflammation associated with upregulation of NF-κB signaling is a common feature of multiple neurodegenerative diseases [3][16]. Thus, the search for selective blockers of NF-κB signaling is an attractive task for both anticancer and anti-neurodegenerative disorders’ drug discovery. In 2003, Tobe et al. synthesized a novel pool of quinazoline derivatives that could act as antagonists of the NF-κB signaling pathway [4][5][8,9]. Analyzing various quinazoline core derivatives, Tobe et al. revealed the most effective compound, EVP4593 (4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine), blocking NF-κB signaling at nanomolar concentrations (IC50 11 nM) [5][9]. However, even almost 20 years later, the specific molecular target for EVP4593 remains unclear. It has been established that the EVP4593 does not directly influence any key protein involved in NF-κB signaling, including protein kinase C (PKC), IκB kinase (IKK) complex and transcriptional factor NF-κB (Figure 1) [6][10]. Therefore, it was assumed that the molecular target of the EVP4593 should be located upstream of NF-κB signaling. The key upstream activators of NF-κB signaling are receptors of interleukins, tumor necrosis factor, lipopolysaccharides and calcium ions [1][14]. It is well-known that calcium influx through store-operated calcium (SOC) channels triggers the activation of the NF-κB cascade [5][6][7][9,10,11] (Figure 1). Moreover, established store-operated calcium entry (SOCE) blockers SKF96365 [8][17] and BTP2 [9][18] were reported to suppress NF-κB activity. The hypothesis that EVP4593 affects SOC channels, thus modulating NF-κB signaling, was proven for the first time in 2011 by Wu et al. [6][10].
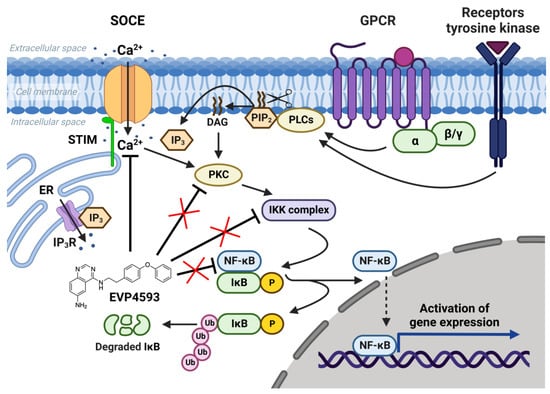
Figure 1. NF-κB signaling pathway and SOCE. SOC channels are activated as a result of intracellular calcium store depletion caused by the activation of inositol-1,4,5-trisphosphate receptor (IP3R). Calcium influx through SOC channels activates protein kinase C (PKC), which activates the IκB kinase (IKK) complex containing the kinases IKKα and IKKβ and the regulatory protein IKKγ. Activated IKK complex phosphorylates inhibitory protein IκB and recruits NF-κB dimers (p50, p52, p65/RelA, RelB and c-Rel). NF-κB free from inhibitory protein IκB migrate to the nucleus and initiate gene expression. EVP4593 does not directly affect any key protein of the NF-κB signal pathway but inhibits SOCE, which is required for NF-κB activation.
2. EVP4593 and SOCE
In 2011, it was shown for the first time that EVP4593 at a concentration of 300 nM can reversibly block SOC channels and reduce pathologically elevated SOCE in an Huntington’s disease (HD) neuroblastoma cell model, expressing full-length mutant huntingtin (138Q), and in striatal neurons isolated from HD-specific YAC128 mice [6][10]. The current–voltage relationships of the registered SOC currents corresponded to the SOC channels formed by the transient receptor potential canonical (TRPC) proteins [6][10]. Moreover, suppression of TRPC1 significantly reduced SOC currents in SK-N-SH cells, modeling HD; at that point, the remaining currents were almost insensitive to further inhibition by EVP4593. Nevertheless, EVP4593 failed to inhibit SOC currents in SK-N-SH cells with overexpressed TRPC1 [6][10]. Therefore, it was concluded that EVP4593 affects heteromeric channels containing TRPC1 as one of the subunits but not homomeric TRPC1 channels. Besides SOC channels’ inhibitory activity, EVP4593 was shown to delay a progression of a motor phenotype in the fly model of HD [6][10]. Thus, SOC channels (especially TRPC1-containing) have become a potential molecular target for EVP4593, and EVP4593 was established as a promising pharmacological substance for HD treatment.
At the same time, the future studies of HD-specific induced pluripotent stem cells (iPSCs)-based GABAergic medium spiny neurons (MSNs) obtained from adult onset HD patients (40–47 glutamine residues in the polyglutamine tract of mutant huntingtin) demonstrated that both Orai- and TRPC-contained SOC channels were sensitive to treatment by 100 nM EVP4593 [10][19]. Thus, it has been supposed that the molecular target for EVP4593 may represent a common regulator of SOC channels, such as STIM1 or STIM2 proteins (Figure 2).
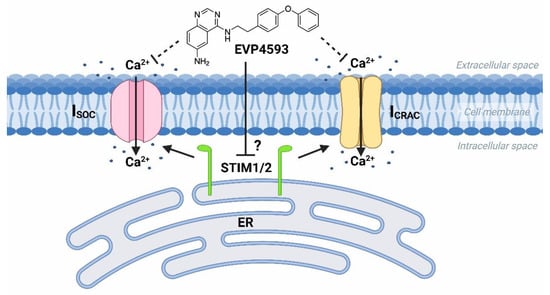
Figure 2. EVP4593 inhibits both Orai- and TRPC-contained store-operated calcium channels. EVP4593 blocks currents through both types of SOC channels that are Orai-contained (ICRAC) and TRPC-contained (ISOC).
Additional experimental data obtained by using fluorescent calcium imaging (the method’s description is available in [11][20]) indicated that EVP4593 could not discriminate between STIM1 and STIM2 in HEK293 cells, affecting SOC in both STIM1 and STIM2 knockout cell lines (Figure 3).
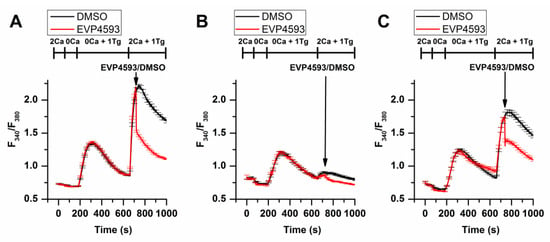
Figure 3. EVP4593 suppresses SOCE in both STIM1 (STIM1KO) and STIM2 (STIM2KO) knockout HEK293 cell lines. Normalized relative fluorescence of Fura-2AM associated with cytosolic calcium level in HEK293 (A), HEK293 STIM1KO (B) and HEK STIM2KO (C) cells during thapsigargin-induced calcium response. The arrow indicates the supply of 1µM EVP4593 or 0.1% DMSO, respectively. The used solutions are indicated above the curves. The curves are plotted as the mean ± SEM.
3. EVP4593 and Mitochondrial Complex I
In addition to the well-studied effects of EVP4593 on SOCE and NF-κB signaling, Robin Krishnathas and colleagues identified mitochondrial complex I as a novel target for EVP4593 [12][21]. EVP4593 has been shown to exhibit strong inhibitory activity against mitochondrial complex I at nanomolar concentration. Molecular docking predictions illustrated that EVP4593 may incorporate into the quinone binding site, thus inhibiting mitochondrial complex I [13][22]. However, EVP4593 targeting mitochondrial complex I did not explain the inhibitory effect on SOCE and NF-κB signaling. Perhaps there are some independent targets for EVP4593. It should be noted that the velocity of the EVP4593 effects detected in experiments does not unequivocally conclude which target is primary from between mitochondrial complex I or SOC channels.
Mitochondrial complex I dysfunctions are also associated with oncogenesis and some neurodegenerative disorders [14][15][16][23,24,25]. Thus, EVP4593 can be successfully used in the treatment of cancer and neurodegenerative diseases, with a suitable overlap in effects on SOCE and mitochondrial complex I.
4. EVP4593 and mTOR Signaling
mTOR (Mechanistic/Mammalian Target of Rapamycin) signaling controls a wide range of processes involved in cellular metabolism and plays a crucial role in autophagy regulation [17][18][26,27]. Aberrant mTOR signaling is associated with a number of pathologies. Hyperactivated mTOR signaling marks some types of cancer and promotes proliferation and tumor progression [17][26]. mTOR signaling inhibitors are considered promising anticancer drugs [19][28]. Indeed, the promising therapeutic strategy is the suppression of the mTOR signaling pathway and, consequently, the enhancement of autophagy [18][27]. Ran Marciano, Manu Prasad and colleagues firstly found that EVP4593 inhibited the mTOR pathway in tumor cells growing under glucose starvation but not under normal conditions [20][29]. This fact indicates the absence of direct interaction of EVP4593 with the components of mTOR signaling. At the same time, low levels of phosphorylated-4EBP1, phosphorylated-P70S6K and phosphorylated-S6RP under EVP4593 treatment indicates inhibition of the mTORC1 pathway [20][29]. Since EVP4593 may inhibit mitochondrial complex I, the tumor cells pretreated by EVP4593 may have a significantly lower level of ATP under glucose deprivation, which leads to an inhibition of the AMPK/mTORC1 pathway and suppression of tumor progression. Additionally, EVP4593 inhibits the NF-ĸB pathway that promotes survival under glucose starvation (as well as other NF-ĸB inhibitors) [21][30]. Thus, EVP4593 promotes the selective death of the most glucose-dependent tumor cells.
The upregulation and involvement of the mTOR signaling pathway in neurodegenerative pathologies, including Huntington’s disease, is excellently discussed in the review by Professor Henry Querfurth [18][27]. The autophagy induction by mTOR signaling antagonists is accompanied by the removal of mutant protein aggregates and may protect neurons from degeneration [18][27]. Moreover, it was also reported that other NF-κB inhibition may reduce the number of autophagosomes [22][32]. Possible controversies can be potentially solved because the reduced number of autophagosomes may indicate the high efficacy of autophagy, whereas a large number of autophagosomes may be a result of autophagy block and the accumulation of “pre-autophagosomes”.
The connection between mTORC1 signaling and SOCE is interesting. The data suggest that mTORC1 signaling is a positive regulator of SOCE [23][24][25][33,34,35]. Additionally, SOCE inhibition led to enhanced autophagy probably through the Akt/mTOR signaling pathway [26][27][36,37]. Furthermore, mTORC1 is a positive regulator of NF-κB signaling [28][38]. mTORC1 may phosphorylate IKKα/β and induce the transcriptional activity of NF-κB [28][38]. Resveratrol, which is known to inhibit NF-κB and mTOR signaling, reduce SOCE in preincubation experiments and also enhance autophagy [29][39]. However, the role of calcium signaling in the regulation of autophagy remains complicated [30][40]. Therefore, for the long-term effect of EVP4593, the following axis can be formed: inhibition of mitochondrial complex I—inhibition of the AMPK/mTORC1 pathway (or Akt/mTOR pathway)—inhibition NF-κB signaling—inhibition of SOCE (Figure 4). Nevertheless, EVP4593 also demonstrates an acute inhibitory SOCE effect confirmed by patch-clamp experiments and fluorescent calcium imaging [6][10][31][32][10,19,31,41] (Figure 3 and Figure 4).
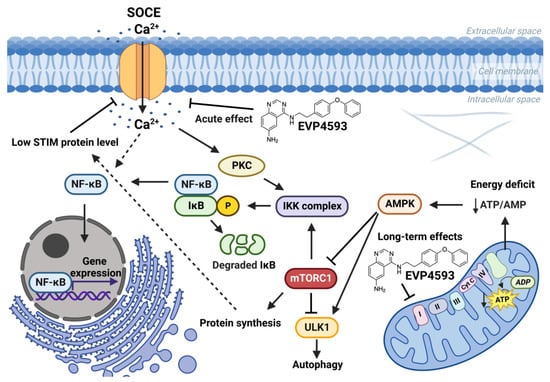
Figure 4. Effects of EVP4593 on cell signaling. EVP4593 has both acute and long-term effects. The acute effect is to directly inhibit SOCE. Long-term effects may be associated with the inhibition of the mitochondrial complex I. Energy deficit induced by EVP4593 inhibits the AMPK/mTORC1 pathway or activates Unc-51 such as autophagy activating kinase 1 (ULK1), which leads to enhanced autophagy and suppressed protein synthesis. Additionally, EVP4593 can negatively affect NF-κB signaling by inhibiting SOCE or mTORC1 signaling. Consequently, EVP4593 also suppresses the expression of NF-κB -dependent genes. The lower levels of STIM proteins due to EVP4593 action may also result in reduced SOCE.
5. EVP4593 and Gene Expression
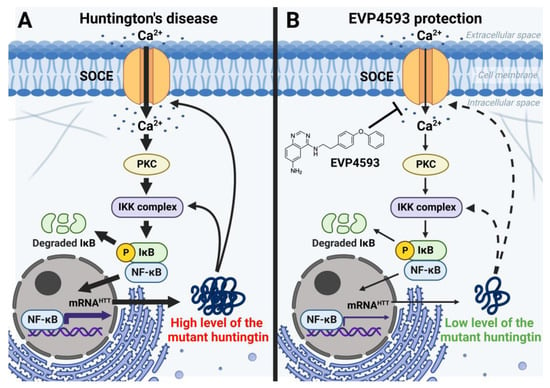
In addition to an acute inhibitory effect of EVP4593 on SOCE, the long-term effects of incubation cells with EVP4593 have also been observed. The modulation of gene expression by EVP4593 is obvious since the expression of many genes depends on NF-κB. Indeed, it was found that incubation of HD-specific MSNs with 300 nM EVP4593 for 24–48 h reduced excessive levels of the huntingtin protein [33][42]. These data were not surprising because the expression of the huntingtin gene depends on NF-κB [34][43] (Figure 5). Notably, modulation of huntingtin expression may have therapeutic applications. It has been shown that even a 10% decrease in mutant huntingtin level has a neuroprotective effect, while even a 90% decrease in normal huntingtin level has no pathological effect [35][44].Figure 5. EVP4593 reduces the level of the huntingtin protein. Huntingtin gene expression is regulated by NF-κB-dependent promoter/enhancer. (A) Elevated SOCE in HD induces hyperactivation of NF-κB signaling, resulting in a high level of the huntingtin protein. Mutant huntingtin enhances calcium influx through SOC channels and potentiates the IKK complex. Thus, a pathological vicious circle is formed. (B) EVP4593 attenuates pathologically enhanced SOCE and decreases NF-κB-dependent huntingtin production.
What is interesting is that proteins, encoded by NF-κB-independent genes, may also change their levels upon treatment by EVP4593. SOC channels’ activator STIM2, which has a high level associated with excessive SOCE in HD-specific neurons [33][42], can also be downregulated by the application of EVP4593, thus protecting neurons from toxic calcium influx. Another research group reported that excessive STIM2-dependent SOC channels’ activity appears to lead to spine loss in YAC128 (HD mice model) MSN [36][45], confirming the key role of STIM2 at a high level in HD pathogenesis and establishing STIM2 as a promising target for anti-HD drugs. On the other hand, it is extremely important that EVP4593 reduces excessive STIM2 levels to control values since it has been reported that downregulation of STIM2 can be dangerous because STIM2-dependent stability of mushroom spines was shown to be a mechanism of hippocampal synaptic loss in a mice model of Alzheimer’s disease [37][46]. Curiously, despite the expression of the STIM2 encoding gene, it does not depend on NF-κB; another blocker of NF-κB signaling, wogonin, can also reduce STIM2 levels [38][47]. Wogonin also inhibits the mTOR pathway and can have a similar effect as rapamycin on the STIM protein level [39][48].