Diabetes mellitus is a chronic endocrine disease, affecting more than 400 million people around the world. Patients with poorly controlled blood glucose levels are liable to suffer from life-threatening complications, such as cardiovascular, neuropathy, retinopathy and even premature death. Today, subcutaneous parenteral is still the most common route for insulin therapy. Oral insulin administration is favourable and convenient to the patients. In contrast to injection route, oral insulin delivery mimics the physiological pathway of endogenous insulin secretion. However, oral insulin has poor bioavailability (less than 2%) due to the harsh physiological environment through the gastrointestinal tract (GIT). Over the last few decades, many attempts have been made to achieve an effective oral insulin formulation with high bioavailability using insulin encapsulation into nanoparticles as advanced technology. Various natural polymers have been employed to fabricate nanoparticles as a delivery vehicle for insulin oral administration. Chitosan, a natural polymer, is extensively studied due to the attractive properties, such as biodegradability, biocompatibility, bioactivity, nontoxicity and polycationic nature. Numerous studies were conducted to evaluate chitosan and chitosan derivatives-based nanoparticles capabilities for oral insulin delivery. This review highlights strategies that have been applied in the recent five years to fabricate chitosan/chitosan derivatives-based nanoparticles for oral insulin delivery. A summary of the barriers hurdle insulin absorption rendering its low bioavailability such as physical, chemical and enzymatic barriers are highlighted with an emphasis on the most common methods of chitosan nanoparticles preparation. Nanocarriers are able to improve the absorption of insulin through GIT, deliver insulin to the blood circulation and lower blood glucose levels. In spite of some drawbacks encountered in this technology, chitosan and chitosan derivatives-based nanoparticles are greatly promising entities for oral insulin delivery.
- Insulin oral delivery
- Chitosan
- Nanoparticles
1. Introduction
Diabetes mellitus (DM), one of the major epidemics worldwide of the 21st century, is a chronic disease that developed in about 451 million people in 2017 and this number is anticipated to increase to 693 million by 2045 worldwide [1,2][1][2]. To date, subcutaneous injections remain the conventional way to deliver insulin daily. However, this route is associated with several drawbacks including poor patient compliance as a result of needle fears, allergic reactions, pain and hypoglycemic episodes [3]. Oral insulin delivery, on the other hand, has been the research of interest globally for decades. Effective oral insulin dose must survive along the gastrointestinal tract (GIT), cross the mucus layer, transport through the intestinal epithelial cells, enter the liver via portal vein and finally reach the systemic circulation [4]. However, mere oral administration of insulin is encountered with enzymatic and physiological barriers that negate insulin absorption through intestinal epithelial cells. Such hurdles render insulin poor oral bioavailability, despite the oral route is the most favourable mode of diabetes management [1].
In order to circumvent the above mentioned challenges, numerous studies have been carried out to develop efficient oral insulin delivery systems where nanotechnology appeared to be a favourable platform. Currently, the application of nanomaterials attracts wider attention in pharmaceutical and biomedical research. Nanoparticulates are defined as entities that are synthesized using nanomaterials that endow unique functionality to the delivery system. The drug content and release profiles of nanosystems are tailorable simply by modulating their starting material composition and physical traits [5]. Generally, nanocarriers can be classified according to their compositional structure into polymeric nanoparticle, lipid-based nanoparticles and inorganic nanoparticle (Figure 1) [6]. In the last two decades, great interest was granted to polymeric and lipid-based nanoparticles over inorganic metal ones for proteins/peptides oral delivery, owing to their biocompatibility and biodegradability, as well as promising clinical outcomes [7,8][7][8]. Polymeric nanoparticles being inert and non-immunogenicity, it enables them to escape from endosomal recognition and avoiding of degradation by lysosomes [6]. Moreover, while nanoparticles generally facilitate insulin transportation in the intestine by both transcellular and paracellular pathways, polymeric nanoparticles significantly enhance insulin absorption through paracellular pathway by reversely opening the tight junctions between adjacent cells [9]. Thus, of all the nanoparticle used in drug delivery designs, polymeric nanoparticles have gained great interest. Furthermore, methods of formulation are widely available therefore, the range of applications has been expanding to include variety of hydrophilic and hydrophobic dugs of chemical drug classes and dosage forms [10,11][10][11]. The smart nanocarriers, synthesized from stimulus-responsive building blocks as part of a polymeric structure, can be controlled to release drugs in response to environmental stimuli such as temperature and pH. In addition, nanocarriers can be decorated with targeting ligand for site-specific drug delivery [12]. Polymeric nanoparticles can be either synthesized from biodegradable synthetic polymers, such as poly(lactide-glycolide) (PLGA) copolymers, polyacrylates, or from natural polymers, such as chitosan, alginate, collagen and albumin. Notable advantages of the natural polymeric-based nanoparticles render them particularly unique due to their abundance in nature, non-toxic with established safety profile and easily modifiable [13].
Among all polysaccharides, chitosan has been the primary interest for many investigators in the designing of oral drug delivery system as a function of its biodegradable, biocompatible, smooth of processing and its digestibility by colonic microbial enzymes to emerge colon-targeted delivery of drugs [14]. Nanoparticles-based chitosan are particularly favourable for the mucosal route due to low toxicity, tunable physiochemical properties and mucoadhesion. There are several methods to formulate chitosan nanoparticles, such as ionic gelation, polyelectrolyte complexation, reverse micellar, emulsion solvent diffusion and electrospraying techniques [15]. Careful selection of nanoparticles composition and method of preparation is essential to meet the objectives of protecting the encapsulant (insulin) and deliver it in a sufficient manner to the blood circulation hence, improve its bioavailability. Thus, the nanoparticle formulator must precisely match the desired chemical and physical attributes of chitosan with reference to the biological environment, with chitosan processing technique [10].
What makes chitosan unique over other polysaccharides for oral drug delivery is its chemical structure that allows specific modifications through modulation in the chitosan amine or hydroxyl functional groups [16]. With regards to pharmaceutical applications, these chemical moieties can be utilised to conjugate drugs directly or via linkers. Abundance of amino groups on the backbone of chitosan would enable any amine related conjugations with other molecules, such as methacrylation [17] and carboxymethylation [18].
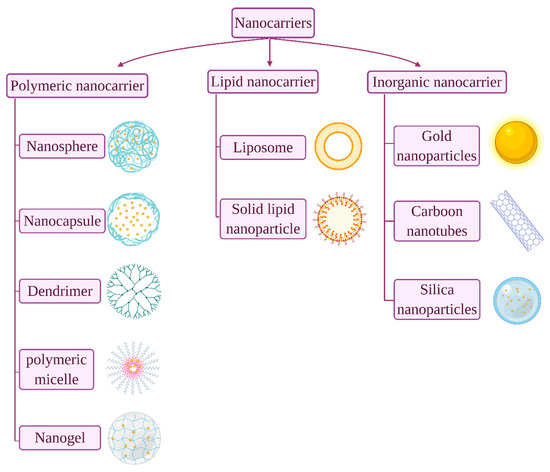
Figure 1. Common nanocarr
Common nanocarriers used for oral protein/peptide delivery.
Thiers used for oral protein/peptide delivery.
Thisentry review will discuss how recent developments in chitosan/chitosan derivatives-based nanotechnology have been emerged in a multitude of platforms for safe and efficient delivery of insulin orally for the treatment of DM.
1.1. Diabetes Mellitus
Diabetes mellitus is a chronic endocrine disease in which an elevation of blood glucose level occurs as a result of reduced or inability of pancreas to produce insulin or due to peripheral tissue uptake defects of insulin [19]. Diabetes can primarily be classified into two types: type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). In T1DM, the pancreas terminates or reduces insulin production due to pancreatic β-cell destruction, whereas in T2DM, the cells manifest low sensitivity towards insulin and consequently both types lead to hyperglycemia [20]. Poorly controlled blood glucose levels can bring about serious adverse effects in cardiovascular system, nervous system, retina, and even early death [21[21][22],22], hence exogenous insulin intake is imperative in patients with T1DM and advanced T2DM [23].
1.2. Insulin
Insulin is an anabolic polypeptide hormone, synthesized in high amounts by islets of Langerhans of pancreatic β-cells and is responsible to maintain blood glucose level at normal ranges. Proinsulin (an insulin prohormone precursor) is composed of three domains: an amino-terminal B chain, a carboxy-terminal A chain, and a connecting peptide in the middle denoted by C-peptide. By the cleavage of C-peptide, insulin is formed as a quaternary macromolecule composed of two polypeptide chains, A chain (21 amino acid residues) and B chain (30 amino acid residues) that are linked by disulphide bonds (Figure 2) [24]. In 1922, insulin was first successfully isolated by a team of Canadian scientists in Toronto; a discovery that brought about a true medical success and a milestone in the history of treating diabetes [25]. While insulin has been available for treating diabetes for almost a century now, to date, the most common insulin therapy is to be administered to diabetic patients through the parenteral route. Even though this route is still the best route in terms of effectiveness, insulin administered subcutaneously is delivered directly to the peripheral circulation, unmet the endogenous insulin pathway [26]. As a result, insulin enters the liver, which is the main target organ of insulin, at a much lower concentration than normal endogenous insulin which may give rise to hyperinsulinemia, weight gain, and hypoglycemic risks [27]. Moreover, injection is invasive and may induce local tissue necrosis, infection, and allergy that in long-term treatment may lead to low patient compliance and serious complications such as, nerve damage, insulin resistance and hypokalemia [28]. Therefore, alternative routes for insulin delivery were widely investigated recently, such as pulmonary, nasal, buccal, transdermal and oral [29].
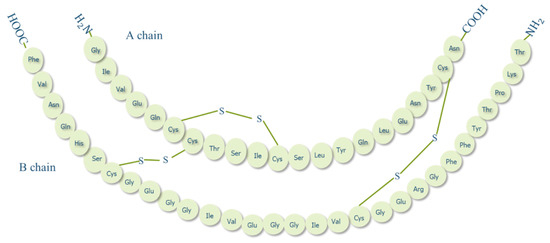
Figure 2. Structure of human insulin.
1.3. Oral Insulin Delivery
Among all alternative routes for insulin administration, oral route is the most favourable approach and mimics the endogenous insulin pathway. After oral administration, insulin is absorbed from intestinal lumen and transported via portal circulation to the liver in which the first pass effect takes place, generating a high porto-systemic gradient (Figure 3). Insulin then reaches peripheral circulation at relatively low levels, imitating physiological insulin pathway and avoiding side effects associated with subcutaneous route such as, hypoglycemic episodes and weight gain [30]. However, insulin oral administration is usually characterised by poor bioavailability (<2%) [31], due to enzymatic degradation, low stability at different pHs and low permeability of GIT [32].
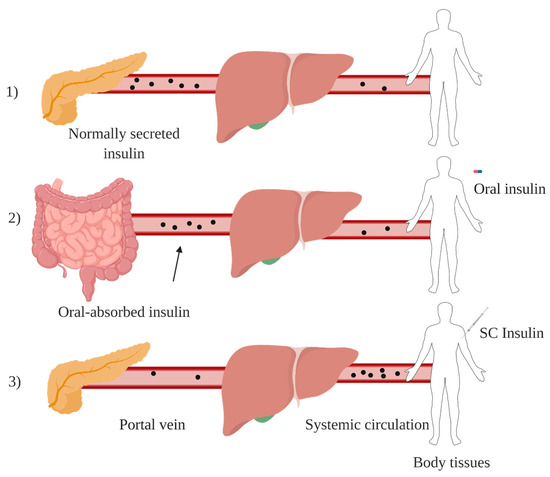
Figure 3. Endogenous insulin pathway under normal physiology (1), orally administered insulin pathway (2) and injected insulin pathway in diabetic patients (3). Created with BioRender.com.
1.4. Barriers to Oral Insulin Delivery
The development of oral insulin delivery system is associated with persistent physiological challenges, where GIT is envisaged to be the main barrier. GIT is responsible for digesting food and selectively absorbing nutrients, electrolytes and fluids. Concurrently, GIT confers a protective barrier against toxic materials such as peptides, viruses and bacteria [33]. These functions are accomplished by a layer of neighboring absorptive and secretory cells, tight junctions that narrowing the paracellular spaces between epithelial cells, a viscous layer of mucus, different pH conditions along GIT and the presence of enzymes involved in food digestion [34]. Table 1 represents a summary of these conditions or barriers that hinder oral delivery of insulin and limit its bioavailability.
Table 1.
Effects of different barriers in gastrointestinal tract (GIT) against insulin oral delivery.
|
Barriers Against Oral Insulin Administration |
||||
|
Physical Barriers |
Chemical Barriers |
Enzymatic Barriers |
||
|
Mucus layer |
Epithelial layer (Trans-cellular transportation) |
Tight junctions (Para-cellular transportation) |
Stomach: highly acidic (pH 1–3.7) ↓ Denaturation and degradation of insulin.
Intestine: neutral and slightly alkaline (pH 6–8).
This variation in pH values may cause pH-induced oxidation and deamination of the protein [35][36]. |
Insulin breakdowns by the protease’s enzymes found in the GIT [24].
Stomach: pepsin.
Intestine: mainly trypsin, chymotrypsin cytosolic and membrane-bound enzymes in the microvilli of intestinal enterocytes [37][38]. |
|
Viscous, hydrophilic, negatively charged layer ↓ Permitting only hydrophilic net-neutral molecules to pass ↓ Hydrophobic drugs and proteins are unable to cross, while cationic compounds exhibit low diffusion rate than neutral ones [31][39]. |
Highly limited to lipophilic drugs with molecular weight less than 700 Da as the membrane is mainly consisting of phospholipid bilayers [30].
Insulin is hydrophilic protein with high molecular weight 5800 Da. |
Regulate the transportation of molecules in between the epithelial cells.
Selectively permeable to small hydrophilic molecules [30][34][40]. |
||
|
Barriers Against Oral Insulin Administration |
||||
|
Physical Barriers |
Chemical Barriers |
Enzymatic Barriers |
||
|
Mucus layer |
Epithelial layer (Trans-cellular transportation) |
Tight junctions (Para-cellular transportation) |
Stomach: highly acidic (pH 1–3.7) ↓ Denaturation and degradation of insulin.
Intestine: neutral and slightly alkaline (pH 6–8).
This variation in pH values may cause pH-induced oxidation and deamination of the protein [37,38]. |
Insulin breakdowns by the protease’s enzymes found in the GIT [24].
Stomach: pepsin.
Intestine: mainly trypsin, chymotrypsin cytosolic and membrane-bound enzymes in the microvilli of intestinal enterocytes [39,40]. |
|
Viscous, hydrophilic, negatively charged layer ↓ Permitting only hydrophilic net-neutral molecules to pass ↓ Hydrophobic drugs and proteins are unable to cross, while cationic compounds exhibit low diffusion rate than neutral ones [31,35]. |
Highly limited to lipophilic drugs with molecular weight less than 700 Da as the membrane is mainly consisting of phospholipid bilayers [30].
Insulin is hydrophilic protein with high molecular weight 5800 Da. |
Regulate the transportation of molecules in between the epithelial cells.
Selectively permeable to small hydrophilic molecules [30,34,36]. |
||
1.5. Chitosan
Chitosan and chitin have received immense attention in different fields in both research and industrial areas, not only because of their biocompatibility, biodegradability and non-toxic properties, but also because they are readily available, inexpensive and environment-friendly biopolymers [41]. Chitin, the second next to cellulose known as the most abundant natural polysaccharides, is a linear polymer comprises of ß-1,4-linked N-acetyl-D-glucosamine, found in the exoskeleton of insects and crustaceans like crab and shrimp, as well as cell walls of fungi [38][36]. Chitin can be found in diverse degrees of acetylation, in which the degree of acetylation (DA) equals 50% or lower, chitin becomes soluble in acidic aqueous solution and is called chitosan [42]. Chitosan, which is the most valuable derivative of chitin, can be obtained by three different chitin-deacetylation methods: chemical alkaline, microbial and enzyme-based method [43]. Structurally, chitosan is a linear polymer composed of randomly distributed β1-4 linked D-glucosamine and N-acetyl D-glucosamine units, with one amino group (NH2) at C-2, and two hydroxyl (OH) groups at C-3 and C-6 in each repeating glycosidic units (Figure 4) [44,45][44][45].
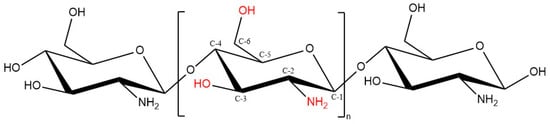
Figure 4. Chitosan chemical structure (DD ≥ 50%).
Chitosan has received widespread interest to deliver therapeutics for different diseases due to its unique attributes including biocompatibility, biodegradability, mucoadhesion, absorption-enhancing effect, low immune reactions, simplicity of functional groups grafting and non-toxicity [46]. Chitosan also possesses diverse biological activities, such as antioxidant [47], antimicrobial [48], anticancer [49] and wound healing propensity [50]. This advocates the intensive research studies of chitosan and its derivatives for various pharmaceutical and medical applications including tissue engineering [51], food technology [52], wound healing [50], gene delivery [53] and textile industry [54]. Besides, it has also been widely employed in various forms in drug delivery systems such as tablets [55], microspheres [56], nanoparticles [57], nanofibers [58], beads [59], films [60], hydrogels [61], conjugates [62] and chitosan-based nanocomposites [63].
Chitosan has an essential polycationic property ascribing to its pKa value of 6.5; in acidic solutions the amino groups (NH2) on the backbone of chitosan get protonated and become positively charged (NH3+) making chitosan soluble in aqueous acidic solutions; whereas in an alkaline environment, the amino groups lose their positive charges and chitosan renders insoluble [64]. The physiochemical properties of chitosan including viscosity, solubility, adsorption on solids, elasticity, tear strength and biofunctional propensity are greatly attributed to the molecular weight (MW) and degree of deacetylation (DD). The average MW and DD of chitosan may range from 50 to over 2000 kD, and from 30% to 95%, respectively, depending on the source of chitosan and its preparation method from chitin. Other processing conditions may also alter the physical characteristics and performance of the final chitosan product, such as the type and concentration of reagents, time and temperature used throughout the synthesis process [63,65][63][65]. Despite of all the functional properties, chitosan poor solubility hinders its applicability as a result of uneven distribution of acetyl groups and aggregates [66]. This intervenes with the biomedical avails of chitosan, especially at near physiological pH 7.4, where chitosan is insoluble and less effective as an absorption enhancing agent [65,67][65][67]. Hence, it is imperative to modulate chitosan solubility via introducing various derivatives to maximise its applications especially in drug design and drug delivery, such as chitosan-based nanoparticles.
1.6. Chitosan Nanoparticles
Nanoparticles are defined as entities with the size range from 10 to 999 nm. Different types of insulin-loaded nanoparticles for oral delivery administration have been formulated using various materials such as lipids, metals, proteins, natural polymers and synthetic polymers, either alone or combined [68]. Nanoparticles are advantageous due to their small size, large surface area to volume ratio by which their retention time to reach the intestinal absorption sites are prolonged, thus improved permeation and bioavailability. This in turn reduces the frequency and doses of encapsulant and enhances patient compliance [69].
Over the last few decades, different natural polymers have been adopted to ease the limitations associated with oral insulin absorption exploiting advanced nanotechnology. Chitosan and its derivatives are considered as an excellent choice and have been widely investigated in oral insulin designs. The involvement of chitosan as insulin carrier is based on the mucoadhesive property and its ability to reversibly open the tight junctions of epithelial cells. These properties are mainly attributed to the positive moieties possessed by chitosan surface amine groups [70].
Nanoparticles prepared from chitosan and/or chitosan derivatives generally possess a positively charged surface. Chitosan can be tailored to meet a specific goal owing to its unique functional groups, making it a polymer with a formidable range of potential applications [10]. Chitosan nanoparticles can be synthesised using different methods including ionic gelation, emulsion solvent diffusion, emulsion-droplet coalescence, polyelectrolyte complexation, reverse micelle formation, complex coacervation or solvent evaporation methods.
2. Insulin-Loaded Chitosan Nanoparticles
Chitosan nanoparticles have been widely used to encapsulate insulin mainly via ionic gelation and polyelectrolyte complexation methods. This is attributed to their simplicity, and ability to produce nanoparticles under mild conditions where heat, organic solvent, or toxic cross-linking stabilizer agents are negated. Moreover, the prepared nanoparticles possess narrow size distribution [67,88][67][71]. Both ionic gelation and polyelectrolyte complexation are classified as electrostatic complexation method mainly based on the cationic nature of chitosan mediated by protonation of its amine groups in acidic medium [67]. When protonation degree of chitosan chains is adequate, it enables reactions with anionic molecules, providing excellent gel-forming properties that impart a unique opportunity for insulin entrapment [89][72]. The main difference between the two methods is the nature of the anionic molecules. Ionic chitosan gelation is mediated by small anionic molecules, such as phosphate, citrate, sulphate, while polyelectrolyte complexation is achieved by anionic macromolecules such as dextran sulphate, alginate, hyaluronic acid and DNA [88,90][71][73]. The latter approach is often referred to as interfacial coacervation or complex coacervation. One of the most studied polyanion with chitosan is alginate, which is a non-toxic, biocompatible and biodegradable, mucoadhesive and non-immunogenic anionic polymer (Figure 9) [91][74].
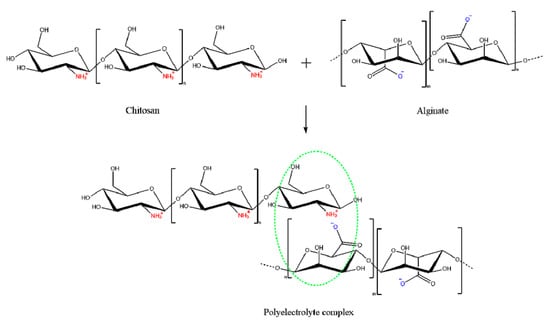
Figure 9. Schematic representation of the interaction between alginate as a polyanion and chitosan as polycation: mixing of the oppositely charged polyelectrolytes leads to formation of a polyelectrolyte complex caused by the electrostatic interactions between the ions.
Chitosan-alginate polyelectrolyte complexation can be enabled via three main approaches: ionotropic pre-gelation of alginate with calcium chloride (CaCl2) or any divalent ions followed by complexation, mixing diluted solutions of chitosan and alginate to acquire a plain complex coacervation, or oil-in-water (o/w) microemulsion of alginate followed by further complexation with chitosan [92][75]. Mukhopadhyay et al., have managed to prepare insulin-loaded core/shell chitosan-alginate nanoparticles using the first approach by admixing dropwise insulin in 0.1 M HCl and CaCl2 to the prepared alginate solution, forming ionic polyelectrolytes after appropriate sonication. This step was followed by chitosan polyelectrolyte complexation by adding chitosan solution with mild stirring to form core-shell nanoparticles via electrostatic interactions. The prepared nanoparticles were characterised by small particle size of 100–200 nm, high encapsulation efficiency of ~85% and pH-responsive sustained release of insulin. The oral administration of insulin-loaded nanoparticles at a dose of 100 IU/kg in the in vivo study exhibited a maximum serum insulin concentration at the 7th h of administration, advocating the nanoparticle’s ability to cross intestinal epithelium and protects insulin from enzymatic degradation in GIT. The results showed that the nanoformulation conferred significantly higher bioavailability (8.11%) than mere oral administration of insulin. Hepatotoxicity test to evaluate any possible toxicity of the nanoparticles by measuring liver-specific enzymes; alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT), has reported that neither liver was damaged nor the liver function was disrupted, demonstrating the safety of the prepared nanoparticles after oral administration [93][76]. Bhattacharyya et al., have applied similar preparation method as above to form core-shell nanocarrier for oral insulin delivery. In their study, instead of using mere alginate, a homogenous blend of polyurethane, a biodegradable and biocompatible synthetic polymer, with alginate (PU-Alg) was utilised to synthesise the core of the desired nanoparticles. The nanoparticles were formulated by adding a mixture solution of insulin and CaCl2 dropwise to PU-Alg blend solution while maintaining sonication for 15 min to allow the construction of the nanoparticle core. Then chitosan solution was added and sonicated for another 15 min to prepare PU-Alg core and chitosan shell nanoparticles. The prepared nanoparticles exhibited small particle size 90–100 nm, more than 90% encapsulation efficiency, prolonged blood glucose lowering in diabetic mice (up to 98 mg/dL for the insulin dose of 100 IU/kg at the 10th h), and relatively improved insulin bioavailability (10.36%) [94][77]. Polyelectrolyte complex of chitosan and alginate has been used by Chen et al., to develop a modified system as a second step after preparing insulin-loaded nanoparticles by double emulsion w/o/w solvent evaporation method. Insulin-loaded w/o/w nanoemulsion of coated chitosan and alginate were firstly prepared, then mixed together for further coacervation of polyelectrolyte complexes as an efficient oral insulin delivery vehicle (Figure 10). The encapsulation efficiency of the obtained alginate-coated and chitosan-coated nanoparticles were 81.5 ± 7.4% and 55.2 ± 7.0%, respectively and average particle size range 200–300 nm. The polyelectrolyte complex exhibited a relative bioavailability of 7.51%, non-cytotoxicity against Caco-2 cell, pH responsive propensity and controlled release profiles (sustained release at pH 6.8, while protecting the drug at pH 1.2) [95][78]. It could be concluded that additional polyelectrolyte complex step has brought about significant improvement on blood glucose lowering propensity by 3-folds for a duration up to 12 h compared to polyelectrolyte complex-free formulation as a result of modulating insulin release throughout the GIT.
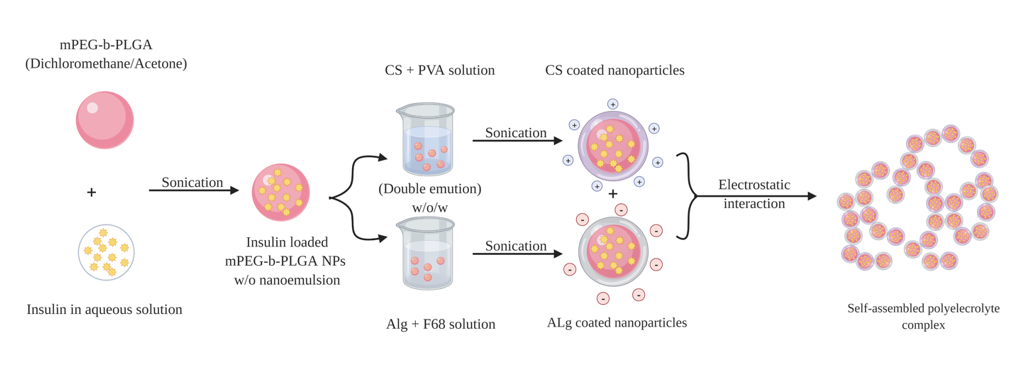
Figure 10. Schematic representation of the double emulsion method used to prepare insulin-loaded polyelectrolyte complexes. Positively charged chitosan nanoparticles (CSNPs) and negatively charged alginate nanoparticles (AlgNPs) are prepared, respectively, then two kinds of nanoparticles are mixed together to self-assemble forming polyelectrolyte complexes by electrostatic interaction.
In another study, low MW PEG (5 kDa) was conjugated with anionic polymer chondroitin sulfate and self-assembled with the cationic chitosan to render negatively charged insulin nanoparticles. In this study, Pereira De Sousa et al., aimed to prepare highly mucus-permeating nanoparticles by combining two different strategies namely, the virus-mimicking and surface PEGylation approaches. Despite of relatively large particle size (510–670 nm) obtained, the nanoparticles exhibited five-fold higher as mucus permeation enhancer as compared to non-PEGylated ones [96][79].
Recent study has utilised Dz13Scr, an anionic oligonucleotide with excellent biocompatibility and minimal cytotoxicity with chitosan to formulate insulin nanoparticles via complex coacervation technique. The developed nanoparticles showed acceptable size range (479 ± 24 nm), uniform polydispersity index (PDI 0.34 ± 0.06) and improved encapsulation efficiency (88.71 ± 0.3%). It was envisaged that this formulation is a potential oral insulin delivery system as it demonstrated improved stability in acidic condition mimicking those in the stomach with only 13% insulin release, while in alkaline medium, a biphasic release pattern of initial burst release (49.49%) followed by a sustained release propensity (88%) in 10 h was attainable. Moreover, the physiochemical properties of the prepared nanoparticles remained stable after being stored for two months at 4 °C as compared to newly-synthesised formulations. The developed nanoparticles achieved a balance between the mucoadhesive property caused by the cationic chitosan and the mucopenetrating capacity attributed to hydrophilic Dz13Scr presence. As a result, the encapsulated insulin was able to permeate across the GI cells (approximately 68% of encapsulated insulin translocated to the basolateral chambers within 1 h) and induces glucose consumption; as it demonstrated comparable effect in promoting the glucose uptake from 16.98% when native insulin used to 20.79% of glucose uptake in C2C12 cells by 12 h of treatment [97][80].
Recently, modified hybrid systems were developed to merge the conventional ionotropic gelation of chitosan with other preparation methods or strategies to obtain the most out of the prepared nanoparticles in terms of preferable characteristics, such as enhanced bioavailability of encapsulant and improved stability along GIT. Erel G et al., have designed insulin-loaded chitosan nanoparticles initially by ionic gelation between chitosan and TPP. As a novel approach, the nanoparticles were then loaded into the inner phase of prepared w/o microemulsion to grant controlled release property, enhance in vivo stability and promote drug absorption in the GIT. The effect of incorporating insulin into chitosan nanoparticle had a significant protective effect. After 8 h of administration of insulin-loaded chitosan nanoparticles embedded in microemulsion, the blood glucose level reduced by 33.6% of the initial blood glucose level, compared to only 17% reduction in the case of nanoparticles-free microemulsion [98][81].
Another new method was developed by He et al., also depends on the electrostatic interaction between chitosan and TPP to prepare size-controlled chitosan nanoparticles for oral insulin delivery called flash nanocomplexation (FNC). In this method, a multi-inlet vortex mixer was used to infuse aqueous solutions of chitosan, TPP and insulin to assure an efficient and rapid mixing to fabricate highly uniform insulin-loaded nanoparticles. This method enables advantage of continuous production of nanoparticles with controlled and reducible particle size (45 nm) while maintaining high encapsulation efficiency (90%), compared with the ordinary dropwise method at 92 ± 8.4 nm and 62.3 ± 4.9% for particle size and encapsulation efficiency, respectively [99][82].
Incorporating chitosan-insulin polyelectrolyte complex (CS-Ins-PEC) with lecithin liposomes to formulate chitosan/lithin liposomal nanovesicles was investigated by Al-Remawi et al., as a possible carrier for insulin oral delivery. Insulin was first reacted with chitosan to form Ins-CS PEC, then the PEC was added to the negatively charged liposomal dispersion developing Ins-CS PEC-associated lecithin liposomes. The optimal formulation possessed high net zeta potential around −30 mV and small particle size of 105 nm ± 17 nm when the ratios of Ins-Cs complex to lecithin was 9% (v/v). The encapsulation efficiency was slightly improved due to the presence of chitosan to interact with insulin comparable to similar chitosan-free formulations [100][83]. For in vivo study, blood glucose lowering effect was observed after 2 h of oral administration accompanied with a prolonged effect up to 8 h. However, the effect was modest that can be attributed to the relatively poor association efficiency [101][84]. Table 2 represents the most recent examples of the compositions, method of preparations and attributes of insulin loaded chitosan-based nanoparticles.
Table 2.
Examples of chitosan-based nanoparticles-loaded insulin.
|
Nanocarrier |
Preparation Method |
Particle Size (nm) |
Zeta Potential (mV) |
Entrapment Efficiency (%) |
In Vitro Insulin Release |
Dose (IU/kg) |
In Vivo Observation |
Reference |
|
Chitosan (CS) MW (25–65 kDa), MW (1.03 × 105 g/mol) |
Polyelectrolyte complexation |
216 |
+3.89 |
78.3 |
A burst release with max. of 26.7% of insulin release was found in pH 1.2, followed by a sustained and prolonged insulin release (79–84%) through 24 h. |
Oral: 50–100 SC: 5 |
Insulin-loaded CS/ALG NPs (50 and 100 IU/kg) showed reduction in the blood glucose level to 143 and 104 mg/dL, respectively, with sustained effect up to 9 h. |
[76] |
|
Medium MW, 75%, 85% deacetylated Chitosan + TPP ratio 6:1 |
Ionic gelation method |
Nanoparticle 356.5 ± 43.4 (Microemultion) 99.1 ± 28.7 |
Nanoparticle 46.5 (Microemultion) 13.1 |
- |
At pH 2.5 after 2 h, insulin release from microemulsion was 48.1%. At pH 6.8 after 2 h, the release was 51.2% and after 3 h it was 66.1%. |
Oral: 50 SC: 1 |
Plasma glucose level reduced to 68.7% after 3 h and it maintained at 66.4% of the initial blood glucose level after 8 h. |
[81] |
|
Chitosan 25 kDa, + Chondroitin sulphate (ChS) 20–30 KDa + Polyethylene glycol 5000 Da (PEG) |
Ionic gelation |
510–670 |
−1 to −5 |
2.18 ± 0.70 |
In simulated intestinal fluid (SIF) buffer, insulin release profile showed a gradual release of the protein reaching 65% in 4 h, followed by a plateau. |
- |
- |
[79] |
|
90 KDa MW, 85% deacetylated chitosan + TPP |
Flash nanocomplexation using multi-inlet vortex mixer |
46.2 ± 2.7 |
9.4 ± 1.2 |
91.0 ± 1.7 |
The amount of released insulin at pH 2.5 was about 16%, while negligible amount at pH 6.6, and a sustained release of insulin within a few hours at pH 7.4 |
Oral: 60 or 120 SC: 10 |
Gradual but distinct reduction of blood glucose levels by 51% (60 IU/kg) and 59% (120 IU/kg) within 8 h. |
[82] |
|
Chitosan (28 kDa) + Lecithin liposomes + L-Arginine |
CS-insulin dispersion (polyelectrolyte complexation) added to lecithin liposomal dispersion |
105 ± 17 |
−30 |
20 |
Insulin was rapidly released in both 0.1 M HCl and phosphate buffer pH 6.8 media and complete release was achieved almost after 30 min. |
Oral: 50 SC: 1 |
A significant effect was observed at 2 h after oral administration as the blood glucose level was reduced by almost 17% of the initial level and the effect was prolonged for up to 8 h. |
[84] |
|
Low MW 50–190 kDa, ≥75.0% deacetylated chitosan + Iota-carrageenan (CMCi) |
Polyelectrolyte complexation method |
613 ± 41 |
52.5 ± 0.5 |
86.9 ± 2.6 |
After 2 h in simulated gastric fluid (SGF), the release of insulin from the nanoparticles was only 4.91% ± 0.24%, while in SIF, the release of insulin was 86.64% ± 2.20%. |
- |
- |
[85] |
|
Chitosan, alloxan monohydrate + Alginate + Polyurethane (PU-ALG/CS NPs) |
Polyelectrolyte complexation method |
90–110 |
38.5 |
90 |
There was a slight insulin release (13.7%) at pH 1.2 up to 1 h, while moderately release (up to 50%) till 10th h in pH 6.8 buffer solution, whereas sustained release of insulin was noticed at pH 7.4 from 11th h, and reached the maximum insulin release after 20th h (98.32%). |
Oral: 50 and 100 SC: 5 |
Blood glucose level was reduced up to 98 mg/dL for the insulin doses of 100 IU/kg, and 131 mg/dL for the 50 IU/kg dose at the 10th h. |
[77] |
|
Chitosan 95% DD + Alginate + Methoxypolyethylene glycol (mPEG, MW 5.0 kDa) + D, L-Lactide (LA) + Glycolide (GA) + Poly (vinyl alcohol)1788 low-viscosity (PVA) + poly (ethylene glycol)-block-poly (propylene glycol)-block-poly (ethylene glycol) (F68, Mw 8.4 kDa) |
Double-emulsion (w/o/w) solvent evaporation method + Polyelectrolyte complexation |
CS NP 224.4 ± 13.8 Alg NP 260.1 ± 17.1 |
CS NP +13.7 ± 1.6 Alg NP −55.7 ± 6.6 |
CS NP 55.2 ± 7.0 Alg NP 81.5 ± 7.4 |
The insulin loaded PEC enabled a slight insulin release (only 13.91%) in SGF (pH 1.2) within the first 4 h. In contrast, rapid rising rate in the first 4 h (38.03%) at the pH 6.8 took place, and the cumulative drug release increased to 51.57% within 10 h, and reached 80.54% after 60 h. |
Oral: 60 SC: 5 |
The blood glucose level decreased after the oral administration of insulin-loaded PEC with the maximal blood glucose reduction of 30% at 8 h, and 20% after 12 h. Insulin concentration in plasma was increased gradually and resulted in a maximum plasma concentration (41.5 ± 4.4 μIU mL−1) at 10 h. |
[78] |
|
Chitosan (95% deacetylated; MW 150 kDa) + Dz13Scr |
Complex coacervation |
534 ± 24 |
14.57 ± 1.1 |
79.96 ± 3.96 |
Only 14.03% of cumulative insulin released at pH 2, while approximately 85% of insulin was released after 10 h at pH 6.8 phosphate buffer solution. |
- |
- |
[80] |
|
Nanocarrier |
Preparation Method |
Particle Size (nm) |
Zeta Potential (mV) |
Entrapment Efficiency (%) |
In Vitro Insulin Release |
Dose (IU/kg) |
In Vivo Observation |
Reference |
|
Chitosan (CS) MW (25–65 kDa), 83–86% Deacetylation Degree(DD) + Alginate (ALG) MW (1.03 × 105 g/mol) |
Polyelectrolyte complexation |
216 |
+3.89 |
78.3 |
A burst release with max. of 26.7% of insulin release was found in pH 1.2, followed by a sustained and prolonged insulin release (79–84%) through 24 h. |
Oral: 50–100 SC: 5 |
Insulin-loaded CS/ALG NPs (50 and 100 IU/kg) showed reduction in the blood glucose level to 143 and 104 mg/dL, respectively, with sustained effect up to 9 h. |
[93] |
|
Medium MW, 75%, 85% deacetylated Chitosan + TPP ratio 6:1 |
Ionic gelation method |
Nanoparticle 356.5 ± 43.4 (Microemultion) 99.1 ± 28.7 |
Nanoparticle 46.5 (Microemultion) 13.1 |
- |
At pH 2.5 after 2 h, insulin release from microemulsion was 48.1%. At pH 6.8 after 2 h, the release was 51.2% and after 3 h it was 66.1%. |
Oral: 50 SC: 1 |
Plasma glucose level reduced to 68.7% after 3 h and it maintained at 66.4% of the initial blood glucose level after 8 h. |
[98] |
|
Chitosan 25 kDa, + Chondroitin sulphate (ChS) 20–30 KDa + Polyethylene glycol 5000 Da (PEG) |
Ionic gelation |
510–670 |
−1 to −5 |
2.18 ± 0.70 |
In simulated intestinal fluid (SIF) buffer, insulin release profile showed a gradual release of the protein reaching 65% in 4 h, followed by a plateau. |
- |
- |
[96] |
|
90 KDa MW, 85% deacetylated chitosan + TPP |
Flash nanocomplexation using multi-inlet vortex mixer |
46.2 ± 2.7 |
9.4 ± 1.2 |
91.0 ± 1.7 |
The amount of released insulin at pH 2.5 was about 16%, while negligible amount at pH 6.6, and a sustained release of insulin within a few hours at pH 7.4 |
Oral: 60 or 120 SC: 10 |
Gradual but distinct reduction of blood glucose levels by 51% (60 IU/kg) and 59% (120 IU/kg) within 8 h. |
[99] |
|
Chitosan (28 kDa) + Lecithin liposomes + L-Arginine |
CS-insulin dispersion (polyelectrolyte complexation) added to lecithin liposomal dispersion |
105 ± 17 |
−30 |
20 |
Insulin was rapidly released in both 0.1 M HCl and phosphate buffer pH 6.8 media and complete release was achieved almost after 30 min. |
Oral: 50 SC: 1 |
A significant effect was observed at 2 h after oral administration as the blood glucose level was reduced by almost 17% of the initial level and the effect was prolonged for up to 8 h. |
[101] |
|
Low MW 50–190 kDa, ≥75.0% deacetylated chitosan + Iota-carrageenan (CMCi) |
Polyelectrolyte complexation method |
613 ± 41 |
52.5 ± 0.5 |
86.9 ± 2.6 |
After 2 h in simulated gastric fluid (SGF), the release of insulin from the nanoparticles was only 4.91% ± 0.24%, while in SIF, the release of insulin was 86.64% ± 2.20%. |
- |
- |
[102] |
|
Chitosan, alloxan monohydrate + Alginate + Polyurethane (PU-ALG/CS NPs) |
Polyelectrolyte complexation method |
90–110 |
38.5 |
90 |
There was a slight insulin release (13.7%) at pH 1.2 up to 1 h, while moderately release (up to 50%) till 10th h in pH 6.8 buffer solution, whereas sustained release of insulin was noticed at pH 7.4 from 11th h, and reached the maximum insulin release after 20th h (98.32%). |
Oral: 50 and 100 SC: 5 |
Blood glucose level was reduced up to 98 mg/dL for the insulin doses of 100 IU/kg, and 131 mg/dL for the 50 IU/kg dose at the 10th h. |
[94] |
|
Chitosan 95% DD + Alginate + Methoxypolyethylene glycol (mPEG, MW 5.0 kDa) + D, L-Lactide (LA) + Glycolide (GA) + Poly (vinyl alcohol)1788 low-viscosity (PVA) + poly (ethylene glycol)-block-poly (propylene glycol)-block-poly (ethylene glycol) (F68, Mw 8.4 kDa) |
Double-emulsion (w/o/w) solvent evaporation method + Polyelectrolyte complexation |
CS NP 224.4 ± 13.8 Alg NP 260.1 ± 17.1 |
CS NP +13.7 ± 1.6 Alg NP −55.7 ± 6.6 |
CS NP 55.2 ± 7.0 Alg NP 81.5 ± 7.4 |
The insulin loaded PEC enabled a slight insulin release (only 13.91%) in SGF (pH 1.2) within the first 4 h. In contrast, rapid rising rate in the first 4 h (38.03%) at the pH 6.8 took place, and the cumulative drug release increased to 51.57% within 10 h, and reached 80.54% after 60 h. |
Oral: 60 SC: 5 |
The blood glucose level decreased after the oral administration of insulin-loaded PEC with the maximal blood glucose reduction of 30% at 8 h, and 20% after 12 h. Insulin concentration in plasma was increased gradually and resulted in a maximum plasma concentration (41.5 ± 4.4 μIU mL−1) at 10 h. |
[95] |
|
Chitosan (95% deacetylated; MW 150 kDa) + Dz13Scr |
Complex coacervation |
534 ± 24 |
14.57 ± 1.1 |
79.96 ± 3.96 |
Only 14.03% of cumulative insulin released at pH 2, while approximately 85% of insulin was released after 10 h at pH 6.8 phosphate buffer solution. |
- |
- |
[97] |
References
83–86% |
|
Deacetylation Degree(DD) + Alginate (ALG) |
- Un Kim, J.; Muhammad Shahbaz, H.; Lee, H.; Kim, T.; Yang, K.; Hoon Roh, Y.; Park, J. Optimization of Phytic Acid-Crosslinked Chitosan Microspheres for Oral Insulin Delivery Using Responsive Surface Methodology. J. Pharm. 2020, 588, 119736, doi:10.1016/j.ijpharm.2020.119736.
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; López Machado, A.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152.
- Yang, Y.; Liu, Y.; Chen, S.; Cheong, K.-L.; Teng, B. Carboxymethyl β-Cyclodextrin Grafted Carboxymethyl Chitosan Hydrogel-Based Microparticles for Oral Insulin Delivery. Polym. 2020, 246, 116617, doi:10.1016/j.carbpol.2020.116617.
- Lopes, M.A.; Abrahim, B.A.; Cabral, L.M.; Rodrigues, C.R.; Seiça, R.M.F.; de Baptista Veiga, F.J.; Ribeiro, A.J. Intestinal Absorption of Insulin Nanoparticles: Contribution of M Cells. Nanotechnol. Biol. Med. 2014, 10, 1139–1151.
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano Based Drug Delivery Systems: Recent Developments and Future Prospects. Nanobiotechnol. 2018, 16, 71, doi:10.1186/s12951-018-0392-8.
- El-Say, K.M.; El-Sawy, H.S. Polymeric Nanoparticles: Promising Platform for Drug Delivery. J. Pharm. 2017, 528, 675–691, doi:10.1016/j.ijpharm.2017.06.052.
- Santalices, I.; Gonella, A.; Torres, D.; Alonso, M.J. Advances on the Formulation of Proteins Using Nanotechnologies. Drug Deliv. Sci. Technol. 2017, 42, 155–180, doi:10.1016/j.jddst.2017.06.018.
- Sadeghi, S.; Lee, W.K.; Kong, S.N.; Shetty, A.; Drum, C.L. Oral Administration of Protein Nanoparticles: An Emerging Route to Disease Treatment. Res. 2020, 158, 104685, doi:10.1016/j.phrs.2020.104685.
- Zhang, T.; Tang, J.Z.; Fei, X.; Li, Y.; Song, Y.; Qian, Z.; Peng, Q. Can Nanoparticles and Nano‒protein Interactions Bring a Bright Future for Insulin Delivery? Acta Pharm. Sin. B 2020, doi:10.1016/j.apsb.2020.08.016.
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53, doi:10.3390/pharmaceutics9040053.
- Gardouh, A.R.; Attia, M.A.; Enan, E.T.; Elbahaie, A.M.; Fouad, R.A.; El-Shafey, M.; Youssef, A.M.; Alomar, S.Y.; Ali, Z.A.-E.; Zaitone, S.A.; et al. Synthesis and Antitumor Activity of Doxycycline Polymeric Nanoparticles: Effect on Tumor Apoptosis in Solid Ehrlich Carcinoma. Molecules 2020, 25, 3230, doi:10.3390/molecules25143230.
- Liao, Z.; Wong, S.W.; Yeo, H.L.; Zhao, Y. Nanocarriers for Cancer Treatment: Clinical Impact and Safety. NanoImpact 2020, 20, 100253, doi:10.1016/j.impact.2020.100253.
- Wong, K.H.; Lu, A.; Chen, X.; Yang, Z. Natural Ingredient-Based Polymeric Nanoparticles for Cancer Treatment. Molecules 2020, 25, 3620, doi:10.3390/molecules25163620.
- Cheng, H.; Huang, S.; Huang, G. Design and Application of Oral Colon Administration System. Enzym. Inhib. Med. Chem. 2019, 34, 1590–1596, doi:10.1080/14756366.2019.1655406.
- Ciro, Y.; Rojas, J.; Alhajj, M.J.; Carabali, G.A.; Salamanca, C.H. Production and Characterization of Chitosan–Polyanion Nanoparticles by Polyelectrolyte Complexation Assisted by High-Intensity Sonication for the Modified Release of Methotrexate. Pharmaceuticals 2020, 13, 11, doi:10.3390/ph13010011.
- Miao, T.; Wang, J.; Zeng, Y.; Liu, G.; Chen, X. Polysaccharide‐based Controlled Release Systems for Therapeutics Delivery and Tissue Engineering: From Bench to Bedside. Sci. 2018, 5, 1700513.
- Diolosà, M.; Donati, I.; Turco, G.; Cadenaro, M.; Di Lenarda, R.; Breschi, L.; Paoletti, S. Use of Methacrylate-Modified Chitosan to Increase the Durability of Dentine Bonding Systems. Biomacromolecules 2014, 15, 4606–4613.
- Taubner, T.; Marounek, M.; Synytsya, A. Preparation and Characterization of Hydrophobic and Hydrophilic Amidated Derivatives of Carboxymethyl Chitosan and Carboxymethyl β-Glucan. J. Biol. Macromol. 2020, 163, 1433–1443, doi:10.1016/j.ijbiomac.2020.07.257.
- Alai, M.S.; Lin, W.J.; Pingale, S.S. Application of Polymeric Nanoparticles and Micelles in Insulin Oral Delivery. Food Drug Anal. 2015, 23, 351–358, doi:10.1016/j.jfda.2015.01.007.
- Adibah, W.N.; Ahmad, W.; Mahmod, H.; Ali, A.M. A Review of Medicinal Plants and Daily Foods Used in Southeast Asia Possessing Antidiabetic Activity. Agrobiotechnol. 2019, 10, 17–35.
- Jin, X.; Zhu, D.D.; Chen, B.Z.; Ashfaq, M.; Guo, X.D. Insulin Delivery Systems Combined with Microneedle Technology. Drug Deliv. Rev. 2018, 127, 119–137, doi:10.1016/j.addr.2018.03.011.
- Nawi, A.; Mamat, M.; Ahmad, W.M. The Factors That Contribute to Diabetes Mellitus in Malaysia: Alternative Linear Regression Model Approach in the Health Field Involving Diabetes Mellitus Data. J. Public Heal. Clin. Sci. 2018, 5, 2289–7577.
- Zhang, Y.; Yu, J.; Kahkoska, A.R.; Wang, J.; Buse, J.B.; Gu, Z. Advances in Transdermal Insulin Delivery. Drug Deliv. Rev. 2019, 139, 51–70, doi:10.1016/j.addr.2018.12.006.
- Wong, C.Y.; Martinez, J.; Dass, C.R. Oral Delivery of Insulin for Treatment of Diabetes: Status Quo, Challenges and Opportunities. Pharm. Pharmacol. 2016, 68, 1093–1108, doi:10.1111/jphp.12607.
- Robinson, S.D.; Safavi-Hemami, H. Insulin as a Weapon. Toxicon 2016, 123, 56–61, doi:10.1016/j.toxicon.2016.10.010.
- Sun, S.; Liang, N.; Hiromitsu Yamamoto, Y.K.; Cui, F.; Yan, P. Ph-Sensitive Poly(Lactide-Co-Glycolide) Nanoparticle Composite Microcapsules for Oral Delivery of Insulin. J. Nanomed. 2015, 10, 3489–3498, doi:10.2147/IJN.S81715.
- Matteucci, E.; Giampietro, O.; Covolan, V.; Giustarini, D.; Fanti, P.; Rossi, R. Insulin Administration: Present Strategies and Future Directions for a Noninvasive (Possibly More Physiological) Delivery. Drug Des. Devel. Ther. 2015, 9, 3109–3118, doi:10.2147/DDDT.S79322.
- Liu, L.; Zhang, Y.; Yu, S.; Zhang, Z.; He, C.; Chen, X. PH- and Amylase-Responsive Carboxymethyl Starch/Poly(2-Isobutyl-Acrylic Acid) Hybrid Microgels as Effective Enteric Carriers for Oral Insulin Delivery. Biomacromolecules 2018, 19, 2123–2136, doi:10.1021/acs.biomac.8b00215.
- Easa, N.; Alany, R.; Carew, M.; Vangala, A. A Review of Non-Invasive Insulin Delivery Systems for Diabetes Therapy in Clinical Trials over the Past Decade. Drug Discov. Today 2018, doi:10.1016/j.drudis.2018.11.010.
- Gedawy, A.; Martinez, J.; Al-Salami, H.; Dass, C.R. Oral Insulin Delivery: Existing Barriers and Current Counter-Strategies. Pharm. Pharmacol. 2018, 70, 197–213, doi:10.1111/jphp.12852.
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the Diffusion Barrier of Mucus and Absorption Barrier of Epithelium by Self-Assembled Nanoparticles for Oral Delivery of Insulin. ACS Nano 2015, 9, 2345–2356, doi:10.1021/acsnano.5b00028.
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Recent Advancements in Oral Administration of Insulin-Loaded Liposomal Drug Delivery Systems for Diabetes Mellitus. J. Pharm. 2018, 549, 201–217, doi:10.1016/j.ijpharm.2018.07.041.
- Odenwald, M.A.; Turner, J.R. The Intestinal Epithelial Barrier: A Therapeutic Target? Rev. Gastroenterol. Hepatol. 2017, 14, 9–21, doi:10.1038/nrgastro.2016.169.
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier : A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 00, 1–14, doi:10.1080/17474124.2017.1343143.
- Araújo, F.; Martins, C.; Azevedo, C.; Sarmento, B. Chemical Modification of Drug Molecules as Strategy to Reduce Interactions with Mucus. Drug Deliv. Rev. 2017, 124, 98–106, doi:10.1016/j.addr.2017.09.020.
- Muheem, A.; Shakeel, F.; Asadullah, M.; Anwar, M.; Mallick, N.; Kumar, G.; Husain, M.; Jalees, F. A Review on the Strategies for Oral Delivery of Proteins and Peptides and Their Clinical Perspectives. Saudi Pharm. J. 2016, 24, 413–428, doi:10.1016/j.jsps.2014.06.004.
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Potential of Insulin Nanoparticle Formulations for Oral Delivery and Diabetes Treatment. Control. Release 2017, 264, 247–275, doi:10.1016/j.jconrel.2017.09.003.
- McClements, D.J. Encapsulation, Protection, and Delivery of Bioactive Proteins and Peptides Using Nanoparticle and Microparticle Systems: A Review. Colloid Interface Sci. 2018, 253, 1–22, doi:10.1016/j.cis.2018.02.002.
- Lundquist, P.; Artursson, P. Oral Absorption of Peptides and Nanoparticles across the Human Intestine: Opportunities, Limitations and Studies in Human Tissues. Drug Deliv. Rev. 2016, 106, 256–276, doi:10.1016/j.addr.2016.07.007.
- Chen, M.; Sonaje, K.; Chen, K.; Sung, H. Biomaterials A Review of the Prospects for Polymeric Nanoparticle Platforms in Oral Insulin Delivery. Biomaterials 2011, 32, 9826–9838, doi:10.1016/j.biomaterials.2011.08.087.
- Sarode, S.; Upadhyay, P.; Khosa, M.A.; Mak, T.; Shakir, A.; Song, S.; Ullah, A. Overview of Wastewater Treatment Methods with Special Focus on Biopolymer Chitin-Chitosan. J. Biol. Macromol. 2019, 121, 1086–1100, doi:10.1016/J.IJBIOMAC.2018.10.089.
- Rinaudo, M. Chitin and Chitosan: Properties and Applications. Polym. Sci. 2006, 31, 603–632, doi:10.1016/j.progpolymsci.2006.06.001.
- Hosseinnejad, M.; Jafari, S.M. Evaluation of Different Factors Affecting Antimicrobial Properties of Chitosan. J. Biol. Macromol. 2016, 85, 467–475, doi:10.1016/J.IJBIOMAC.2016.01.022.
- Nurhayati, Y.; Manaf, A.A.; Osman, H.; Bakar, A.; Abdullah, C.; Yew, J.; Tang, H. Malaysian Journal of Applied Sciences Effect of Chitosan Oligosaccharides on the Growth of Bifidobacterium Species. J. Appl. Sci. 2016, 1, 13–23.
- Li, L.; Yang, L.; Li, M.; Zhang, L. A Cell-Penetrating Peptide Mediated Chitosan Nanocarriers for Improving Intestinal Insulin Delivery. Polym. 2017, 174, 182–189, doi:10.1016/j.carbpol.2017.06.061.
- Bravo-Anaya, L.M.; Fernández-Solís, K.G.; Rosselgong, J.; Nano-Rodríguez, J.L.E.; Carvajal, F.; Rinaudo, M. Chitosan-DNA Polyelectrolyte Complex: Influence of Chitosan Characteristics and Mechanism of Complex Formation. J. Biol. Macromol. 2019, doi:10.1016/J.IJBIOMAC.2019.01.008.
- Jangra, A.; Lukhi, M.M.; Sulakhiya, K.; Baruah, C.C.; Lahkar, M. Protective Effect of Mangiferin against Lipopolysaccharide-Induced Depressive and Anxiety-like Behaviour in Mice. J. Pharmacol. 2014, 740, 337–345, doi:10.1016/j.ejphar.2014.07.031.
- Chang, A.K.T.; Frias, R.R.; Alvarez, L.V.; Bigol, U.G.; Guzman, J.P.M.D. Comparative Antibacterial Activity of Commercial Chitosan and Chitosan Extracted from Auricularia Sp. Agric. Biotechnol. 2019, 17, 189–195, doi:10.1016/J.BCAB.2018.11.016.
- Gibot, L.; Chabaud, S.; Bouhout, S.; Bolduc, S.; Auger, F.A.; Moulin, V.J. Anticancer Properties of Chitosan on Human Melanoma Are Cell Line Dependent. J. Biol. Macromol. 2015, 72, 370–379, doi:10.1016/J.IJBIOMAC.2014.08.033.
- Miguel, S.P.; Moreira, A.F.; Correia, I.J. Chitosan Based-Asymmetric Membranes for Wound Healing: A Review. J. Biol. Macromol. 2019, 127, 460–475, doi:10.1016/J.IJBIOMAC.2019.01.072.
- Ahsan, S.M.; Thomas, M.; Reddy, K.K.; Sooraparaju, S.G.; Asthana, A.; Bhatnagar, I. Chitosan as Biomaterial in Drug Delivery and Tissue Engineering. J. Biol. Macromol. 2018, 110, 97–109, doi:10.1016/J.IJBIOMAC.2017.08.140.
- Mujtaba, M.; Morsi, R.E.; Kerch, G.; Elsabee, M.Z.; Kaya, M.; Labidi, J.; Khawar, K.M. Current Advancements in Chitosan-Based Film Production for Food Technology; A Review. J. Biol. Macromol. 2019, 121, 889–904, doi:10.1016/J.IJBIOMAC.2018.10.109.
- Baghdan, E.; Pinnapireddy, S.R.; Strehlow, B.; Engelhardt, K.H.; Schäfer, J.; Bakowsky, U. Lipid Coated Chitosan-DNA Nanoparticles for Enhanced Gene Delivery. J. Pharm. 2018, 535, 473–479, doi:10.1016/J.IJPHARM.2017.11.045.
- Shahid-ul-Islam; Butola, B.S. Recent Advances in Chitosan Polysaccharide and Its Derivatives in Antimicrobial Modification of Textile Materials. J. Biol. Macromol. 2019, 121, 905–912, doi:10.1016/J.IJBIOMAC.2018.10.102.
- Khlibsuwan, R.; Pongjanyakul, T. Chitosan-Clay Matrix Tablets for Sustained-Release Drug Delivery: Effect of Chitosan Molecular Weight and Lubricant. Drug Deliv. Sci. Technol. 2016, 35, 303–313, doi:10.1016/j.jddst.2016.08.003.
- He, T.; Wang, W.; Chen, B.; Wang, J.; Liang, Q.; Chen, B. 5-Fluorouracil Monodispersed Chitosan Microspheres: Microfluidic Chip Fabrication with Crosslinking, Characterization, Drug Release and Anticancer Activity. Polym. 2020, 236, 116094, doi:10.1016/j.carbpol.2020.116094.
- El-Alfy, E.A.; El-Bisi, M.K.; Taha, G.M.; Ibrahim, H.M. Preparation of Biocompatible Chitosan Nanoparticles Loaded by Tetracycline, Gentamycin and Ciprofloxacin as Novel Drug Delivery System for Improvement the Antibacterial Properties of Cellulose Based Fabrics. J. Biol. Macromol. 2020, 161, 1247–1260, doi:10.1016/j.ijbiomac.2020.06.118.
- Stie, M.B.; Gätke, J.R.; Wan, F.; Chronakis, I.S.; Jacobsen, J.; Nielsen, H.M. Swelling of Mucoadhesive Electrospun Chitosan/Polyethylene Oxide Nanofibers Facilitates Adhesion to the Sublingual Mucosa. Polym. 2020, 242, 116428, doi:10.1016/j.carbpol.2020.116428.
- De Oliveira, R.L.; da Silva, M.F.; da Silva, S.P.; Cavalcanti, J.V.F.L.; Converti, A.; Porto, T.S. Immobilization of a Commercial Aspergillus Aculeatus Enzyme Preparation with Fructosyltransferase Activity in Chitosan Beads: A Kinetic/Thermodynamic Study and Fructo-Oligosaccharides Continuous Production in Enzymatic Reactor. Food Bioprod. Process. 2020, 122, 169–182, doi:10.1016/j.fbp.2020.05.001.
- da Silva, T.N.; Reynaud, F.; de Souza Picciani, P.H.; de Holanda e Silva, K.G.; Barradas, T.N. Chitosan-Based Films Containing Nanoemulsions of Methyl Salicylate: Formulation Development, Physical-Chemical and in Vitro Drug Release Characterization. J. Biol. Macromol. 2020, 164, 2558–2568, doi:10.1016/j.ijbiomac.2020.08.117.
- Dehghan-Baniani, D.; Chen, Y.; Wang, D.; Bagheri, R.; Solouk, A.; Wu, H. Injectable in Situ Forming Kartogenin-Loaded Chitosan Hydrogel with Tunable Rheological Properties for Cartilage Tissue Engineering. Colloids Surfaces B Biointerfaces 2020, 192, 111059, doi:10.1016/j.colsurfb.2020.111059.
- Tian, L.; Singh, A.; Singh, A.V. Synthesis and Characterization of Pectin-Chitosan Conjugate for Biomedical Application. J. Biol. Macromol. 2020, 153, 533–538, doi:10.1016/j.ijbiomac.2020.02.313.
- Ali, A.; Ahmed, S. A Review on Chitosan and Its Nanocomposites in Drug Delivery. J. Biol. Macromol. 2018, 109, 273–286, doi:10.1016/J.IJBIOMAC.2017.12.078.
- Situ, W.; Xiang, T.; Liang, Y. Chitosan-Based Particles for Protection of Proteins during Storage and Oral Administration. J. Biol. Macromol. 2018, 117, 308–314, doi:10.1016/J.IJBIOMAC.2018.05.208.
- Bakshi, P.S.; Selvakumar, D.; Kadirvelu, K.; Kumar, N.S. Chitosan as an Environment Friendly Biomaterial – a Review on Recent Modifications and Applications. J. Biol. Macromol. 2020, 150, 1072–1083, doi:10.1016/j.ijbiomac.2019.10.113.
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An Update on Potential Biomedical and Pharmaceutical Applications. Drugs 2015, 13, 5156–5186, doi:10.3390/md13085156.
- Ahmed, T.A.; Aljaeid, B.M. Preparation, Characterization, and Potential Application of Chitosan, Chitosan Derivatives, and Chitosan Metal Nanoparticles in Pharmaceutical Drug Delivery. Drug Des. Devel. Ther. 2016, 10, 483–507, doi:10.2147/DDDT.S99651.
- Castro, P.M.; Raquel, A.; Sarmento, B.; Pintado, M. Recent Insights in the Use of Nanocarriers for the Oral Delivery of Bioactive Proteins and Peptides. Peptides 2018, doi:10.1016/j.peptides.2018.01.002.
- Rizvi, S.A.A.; Saleh, A.M. Applications of Nanoparticle Systems in Drug Delivery Technology. Saudi Pharm. J. 2017, 26, 64–70, doi:10.1016/j.jsps.2017.10.012.
- Luo, Y.Y.; Xiong, X.Y.; Tian, Y.; Li, Z.L.; Gong, Y.C.; Li, Y.P. A Review of Biodegradable Polymeric Systems for Oral Insulin Delivery. Drug Deliv. 2015, 23, 1882–1891, doi:10.3109/10717544.2015.1052863.
References
- Un Kim, J.; Muhammad Shahbaz, H.; Lee, H.; Kim, T.; Yang, K.; Hoon Roh, Y.; Park, J. Optimization of Phytic Acid-Crosslinked Chitosan Microspheres for Oral Insulin Delivery Using Responsive Surface Methodology. J. Pharm. 2020, 588, 119736, doi:10.1016/j.ijpharm.2020.119736.
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; López Machado, A.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152.
- Yang, Y.; Liu, Y.; Chen, S.; Cheong, K.-L.; Teng, B. Carboxymethyl β-Cyclodextrin Grafted Carboxymethyl Chitosan Hydrogel-Based Microparticles for Oral Insulin Delivery. Polym. 2020, 246, 116617, doi:10.1016/j.carbpol.2020.116617.
- Lopes, M.A.; Abrahim, B.A.; Cabral, L.M.; Rodrigues, C.R.; Seiça, R.M.F.; de Baptista Veiga, F.J.; Ribeiro, A.J. Intestinal Absorption of Insulin Nanoparticles: Contribution of M Cells. Nanotechnol. Biol. Med. 2014, 10, 1139–1151.
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano Based Drug Delivery Systems: Recent Developments and Future Prospects. Nanobiotechnol. 2018, 16, 71, doi:10.1186/s12951-018-0392-8.
- El-Say, K.M.; El-Sawy, H.S. Polymeric Nanoparticles: Promising Platform for Drug Delivery. J. Pharm. 2017, 528, 675–691, doi:10.1016/j.ijpharm.2017.06.052.
- Santalices, I.; Gonella, A.; Torres, D.; Alonso, M.J. Advances on the Formulation of Proteins Using Nanotechnologies. Drug Deliv. Sci. Technol. 2017, 42, 155–180, doi:10.1016/j.jddst.2017.06.018.
- Sadeghi, S.; Lee, W.K.; Kong, S.N.; Shetty, A.; Drum, C.L. Oral Administration of Protein Nanoparticles: An Emerging Route to Disease Treatment. Res. 2020, 158, 104685, doi:10.1016/j.phrs.2020.104685.
- Zhang, T.; Tang, J.Z.; Fei, X.; Li, Y.; Song, Y.; Qian, Z.; Peng, Q. Can Nanoparticles and Nano‒protein Interactions Bring a Bright Future for Insulin Delivery? Acta Pharm. Sin. B 2020, doi:10.1016/j.apsb.2020.08.016.
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53, doi:10.3390/pharmaceutics9040053.
- Gardouh, A.R.; Attia, M.A.; Enan, E.T.; Elbahaie, A.M.; Fouad, R.A.; El-Shafey, M.; Youssef, A.M.; Alomar, S.Y.; Ali, Z.A.-E.; Zaitone, S.A.; et al. Synthesis and Antitumor Activity of Doxycycline Polymeric Nanoparticles: Effect on Tumor Apoptosis in Solid Ehrlich Carcinoma. Molecules 2020, 25, 3230, doi:10.3390/molecules25143230.
- Liao, Z.; Wong, S.W.; Yeo, H.L.; Zhao, Y. Nanocarriers for Cancer Treatment: Clinical Impact and Safety. NanoImpact 2020, 20, 100253, doi:10.1016/j.impact.2020.100253.
- Wong, K.H.; Lu, A.; Chen, X.; Yang, Z. Natural Ingredient-Based Polymeric Nanoparticles for Cancer Treatment. Molecules 2020, 25, 3620, doi:10.3390/molecules25163620.
- Cheng, H.; Huang, S.; Huang, G. Design and Application of Oral Colon Administration System. Enzym. Inhib. Med. Chem. 2019, 34, 1590–1596, doi:10.1080/14756366.2019.1655406.
- Ciro, Y.; Rojas, J.; Alhajj, M.J.; Carabali, G.A.; Salamanca, C.H. Production and Characterization of Chitosan–Polyanion Nanoparticles by Polyelectrolyte Complexation Assisted by High-Intensity Sonication for the Modified Release of Methotrexate. Pharmaceuticals 2020, 13, 11, doi:10.3390/ph13010011.
- Miao, T.; Wang, J.; Zeng, Y.; Liu, G.; Chen, X. Polysaccharide‐based Controlled Release Systems for Therapeutics Delivery and Tissue Engineering: From Bench to Bedside. Sci. 2018, 5, 1700513.
- Diolosà, M.; Donati, I.; Turco, G.; Cadenaro, M.; Di Lenarda, R.; Breschi, L.; Paoletti, S. Use of Methacrylate-Modified Chitosan to Increase the Durability of Dentine Bonding Systems. Biomacromolecules 2014, 15, 4606–4613.
- Taubner, T.; Marounek, M.; Synytsya, A. Preparation and Characterization of Hydrophobic and Hydrophilic Amidated Derivatives of Carboxymethyl Chitosan and Carboxymethyl β-Glucan. J. Biol. Macromol. 2020, 163, 1433–1443, doi:10.1016/j.ijbiomac.2020.07.257.
- Alai, M.S.; Lin, W.J.; Pingale, S.S. Application of Polymeric Nanoparticles and Micelles in Insulin Oral Delivery. Food Drug Anal. 2015, 23, 351–358, doi:10.1016/j.jfda.2015.01.007.
- Adibah, W.N.; Ahmad, W.; Mahmod, H.; Ali, A.M. A Review of Medicinal Plants and Daily Foods Used in Southeast Asia Possessing Antidiabetic Activity. Agrobiotechnol. 2019, 10, 17–35.
- Jin, X.; Zhu, D.D.; Chen, B.Z.; Ashfaq, M.; Guo, X.D. Insulin Delivery Systems Combined with Microneedle Technology. Drug Deliv. Rev. 2018, 127, 119–137, doi:10.1016/j.addr.2018.03.011.
- Nawi, A.; Mamat, M.; Ahmad, W.M. The Factors That Contribute to Diabetes Mellitus in Malaysia: Alternative Linear Regression Model Approach in the Health Field Involving Diabetes Mellitus Data. J. Public Heal. Clin. Sci. 2018, 5, 2289–7577.
- Zhang, Y.; Yu, J.; Kahkoska, A.R.; Wang, J.; Buse, J.B.; Gu, Z. Advances in Transdermal Insulin Delivery. Drug Deliv. Rev. 2019, 139, 51–70, doi:10.1016/j.addr.2018.12.006.
- Wong, C.Y.; Martinez, J.; Dass, C.R. Oral Delivery of Insulin for Treatment of Diabetes: Status Quo, Challenges and Opportunities. Pharm. Pharmacol. 2016, 68, 1093–1108, doi:10.1111/jphp.12607.
- Robinson, S.D.; Safavi-Hemami, H. Insulin as a Weapon. Toxicon 2016, 123, 56–61, doi:10.1016/j.toxicon.2016.10.010.
- Sun, S.; Liang, N.; Hiromitsu Yamamoto, Y.K.; Cui, F.; Yan, P. Ph-Sensitive Poly(Lactide-Co-Glycolide) Nanoparticle Composite Microcapsules for Oral Delivery of Insulin. J. Nanomed. 2015, 10, 3489–3498, doi:10.2147/IJN.S81715.
- Matteucci, E.; Giampietro, O.; Covolan, V.; Giustarini, D.; Fanti, P.; Rossi, R. Insulin Administration: Present Strategies and Future Directions for a Noninvasive (Possibly More Physiological) Delivery. Drug Des. Devel. Ther. 2015, 9, 3109–3118, doi:10.2147/DDDT.S79322.
- Liu, L.; Zhang, Y.; Yu, S.; Zhang, Z.; He, C.; Chen, X. PH- and Amylase-Responsive Carboxymethyl Starch/Poly(2-Isobutyl-Acrylic Acid) Hybrid Microgels as Effective Enteric Carriers for Oral Insulin Delivery. Biomacromolecules 2018, 19, 2123–2136, doi:10.1021/acs.biomac.8b00215.
- Easa, N.; Alany, R.; Carew, M.; Vangala, A. A Review of Non-Invasive Insulin Delivery Systems for Diabetes Therapy in Clinical Trials over the Past Decade. Drug Discov. Today 2018, doi:10.1016/j.drudis.2018.11.010.
- Gedawy, A.; Martinez, J.; Al-Salami, H.; Dass, C.R. Oral Insulin Delivery: Existing Barriers and Current Counter-Strategies. Pharm. Pharmacol. 2018, 70, 197–213, doi:10.1111/jphp.12852.
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the Diffusion Barrier of Mucus and Absorption Barrier of Epithelium by Self-Assembled Nanoparticles for Oral Delivery of Insulin. ACS Nano 2015, 9, 2345–2356, doi:10.1021/acsnano.5b00028.
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Recent Advancements in Oral Administration of Insulin-Loaded Liposomal Drug Delivery Systems for Diabetes Mellitus. J. Pharm. 2018, 549, 201–217, doi:10.1016/j.ijpharm.2018.07.041.
- Odenwald, M.A.; Turner, J.R. The Intestinal Epithelial Barrier: A Therapeutic Target? Rev. Gastroenterol. Hepatol. 2017, 14, 9–21, doi:10.1038/nrgastro.2016.169.
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier : A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 00, 1–14, doi:10.1080/17474124.2017.1343143.
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Potential of Insulin Nanoparticle Formulations for Oral Delivery and Diabetes Treatment. Control. Release 2017, 264, 247–275, doi:10.1016/j.jconrel.2017.09.003.
- McClements, D.J. Encapsulation, Protection, and Delivery of Bioactive Proteins and Peptides Using Nanoparticle and Microparticle Systems: A Review. Colloid Interface Sci. 2018, 253, 1–22, doi:10.1016/j.cis.2018.02.002.
- Lundquist, P.; Artursson, P. Oral Absorption of Peptides and Nanoparticles across the Human Intestine: Opportunities, Limitations and Studies in Human Tissues. Drug Deliv. Rev. 2016, 106, 256–276, doi:10.1016/j.addr.2016.07.007.
- Chen, M.; Sonaje, K.; Chen, K.; Sung, H. Biomaterials A Review of the Prospects for Polymeric Nanoparticle Platforms in Oral Insulin Delivery. Biomaterials 2011, 32, 9826–9838, doi:10.1016/j.biomaterials.2011.08.087.
- Araújo, F.; Martins, C.; Azevedo, C.; Sarmento, B. Chemical Modification of Drug Molecules as Strategy to Reduce Interactions with Mucus. Drug Deliv. Rev. 2017, 124, 98–106, doi:10.1016/j.addr.2017.09.020.
- Muheem, A.; Shakeel, F.; Asadullah, M.; Anwar, M.; Mallick, N.; Kumar, G.; Husain, M.; Jalees, F. A Review on the Strategies for Oral Delivery of Proteins and Peptides and Their Clinical Perspectives. Saudi Pharm. J. 2016, 24, 413–428, doi:10.1016/j.jsps.2014.06.004.
- Sarode, S.; Upadhyay, P.; Khosa, M.A.; Mak, T.; Shakir, A.; Song, S.; Ullah, A. Overview of Wastewater Treatment Methods with Special Focus on Biopolymer Chitin-Chitosan. J. Biol. Macromol. 2019, 121, 1086–1100, doi:10.1016/J.IJBIOMAC.2018.10.089.
- Rinaudo, M. Chitin and Chitosan: Properties and Applications. Polym. Sci. 2006, 31, 603–632, doi:10.1016/j.progpolymsci.2006.06.001.
- Hosseinnejad, M.; Jafari, S.M. Evaluation of Different Factors Affecting Antimicrobial Properties of Chitosan. J. Biol. Macromol. 2016, 85, 467–475, doi:10.1016/J.IJBIOMAC.2016.01.022.
- Nurhayati, Y.; Manaf, A.A.; Osman, H.; Bakar, A.; Abdullah, C.; Yew, J.; Tang, H. Malaysian Journal of Applied Sciences Effect of Chitosan Oligosaccharides on the Growth of Bifidobacterium Species. J. Appl. Sci. 2016, 1, 13–23.
- Li, L.; Yang, L.; Li, M.; Zhang, L. A Cell-Penetrating Peptide Mediated Chitosan Nanocarriers for Improving Intestinal Insulin Delivery. Polym. 2017, 174, 182–189, doi:10.1016/j.carbpol.2017.06.061.
- Bravo-Anaya, L.M.; Fernández-Solís, K.G.; Rosselgong, J.; Nano-Rodríguez, J.L.E.; Carvajal, F.; Rinaudo, M. Chitosan-DNA Polyelectrolyte Complex: Influence of Chitosan Characteristics and Mechanism of Complex Formation. J. Biol. Macromol. 2019, doi:10.1016/J.IJBIOMAC.2019.01.008.
- Jangra, A.; Lukhi, M.M.; Sulakhiya, K.; Baruah, C.C.; Lahkar, M. Protective Effect of Mangiferin against Lipopolysaccharide-Induced Depressive and Anxiety-like Behaviour in Mice. J. Pharmacol. 2014, 740, 337–345, doi:10.1016/j.ejphar.2014.07.031.
- Chang, A.K.T.; Frias, R.R.; Alvarez, L.V.; Bigol, U.G.; Guzman, J.P.M.D. Comparative Antibacterial Activity of Commercial Chitosan and Chitosan Extracted from Auricularia Sp. Agric. Biotechnol. 2019, 17, 189–195, doi:10.1016/J.BCAB.2018.11.016.
- Gibot, L.; Chabaud, S.; Bouhout, S.; Bolduc, S.; Auger, F.A.; Moulin, V.J. Anticancer Properties of Chitosan on Human Melanoma Are Cell Line Dependent. J. Biol. Macromol. 2015, 72, 370–379, doi:10.1016/J.IJBIOMAC.2014.08.033.
- Miguel, S.P.; Moreira, A.F.; Correia, I.J. Chitosan Based-Asymmetric Membranes for Wound Healing: A Review. J. Biol. Macromol. 2019, 127, 460–475, doi:10.1016/J.IJBIOMAC.2019.01.072.
- Ahsan, S.M.; Thomas, M.; Reddy, K.K.; Sooraparaju, S.G.; Asthana, A.; Bhatnagar, I. Chitosan as Biomaterial in Drug Delivery and Tissue Engineering. J. Biol. Macromol. 2018, 110, 97–109, doi:10.1016/J.IJBIOMAC.2017.08.140.
- Mujtaba, M.; Morsi, R.E.; Kerch, G.; Elsabee, M.Z.; Kaya, M.; Labidi, J.; Khawar, K.M. Current Advancements in Chitosan-Based Film Production for Food Technology; A Review. J. Biol. Macromol. 2019, 121, 889–904, doi:10.1016/J.IJBIOMAC.2018.10.109.
- Baghdan, E.; Pinnapireddy, S.R.; Strehlow, B.; Engelhardt, K.H.; Schäfer, J.; Bakowsky, U. Lipid Coated Chitosan-DNA Nanoparticles for Enhanced Gene Delivery. J. Pharm. 2018, 535, 473–479, doi:10.1016/J.IJPHARM.2017.11.045.
- Shahid-ul-Islam; Butola, B.S. Recent Advances in Chitosan Polysaccharide and Its Derivatives in Antimicrobial Modification of Textile Materials. J. Biol. Macromol. 2019, 121, 905–912, doi:10.1016/J.IJBIOMAC.2018.10.102.
- Khlibsuwan, R.; Pongjanyakul, T. Chitosan-Clay Matrix Tablets for Sustained-Release Drug Delivery: Effect of Chitosan Molecular Weight and Lubricant. Drug Deliv. Sci. Technol. 2016, 35, 303–313, doi:10.1016/j.jddst.2016.08.003.
- He, T.; Wang, W.; Chen, B.; Wang, J.; Liang, Q.; Chen, B. 5-Fluorouracil Monodispersed Chitosan Microspheres: Microfluidic Chip Fabrication with Crosslinking, Characterization, Drug Release and Anticancer Activity. Polym. 2020, 236, 116094, doi:10.1016/j.carbpol.2020.116094.
- El-Alfy, E.A.; El-Bisi, M.K.; Taha, G.M.; Ibrahim, H.M. Preparation of Biocompatible Chitosan Nanoparticles Loaded by Tetracycline, Gentamycin and Ciprofloxacin as Novel Drug Delivery System for Improvement the Antibacterial Properties of Cellulose Based Fabrics. J. Biol. Macromol. 2020, 161, 1247–1260, doi:10.1016/j.ijbiomac.2020.06.118.
- Stie, M.B.; Gätke, J.R.; Wan, F.; Chronakis, I.S.; Jacobsen, J.; Nielsen, H.M. Swelling of Mucoadhesive Electrospun Chitosan/Polyethylene Oxide Nanofibers Facilitates Adhesion to the Sublingual Mucosa. Polym. 2020, 242, 116428, doi:10.1016/j.carbpol.2020.116428.
- De Oliveira, R.L.; da Silva, M.F.; da Silva, S.P.; Cavalcanti, J.V.F.L.; Converti, A.; Porto, T.S. Immobilization of a Commercial Aspergillus Aculeatus Enzyme Preparation with Fructosyltransferase Activity in Chitosan Beads: A Kinetic/Thermodynamic Study and Fructo-Oligosaccharides Continuous Production in Enzymatic Reactor. Food Bioprod. Process. 2020, 122, 169–182, doi:10.1016/j.fbp.2020.05.001.
- da Silva, T.N.; Reynaud, F.; de Souza Picciani, P.H.; de Holanda e Silva, K.G.; Barradas, T.N. Chitosan-Based Films Containing Nanoemulsions of Methyl Salicylate: Formulation Development, Physical-Chemical and in Vitro Drug Release Characterization. J. Biol. Macromol. 2020, 164, 2558–2568, doi:10.1016/j.ijbiomac.2020.08.117.
- Dehghan-Baniani, D.; Chen, Y.; Wang, D.; Bagheri, R.; Solouk, A.; Wu, H. Injectable in Situ Forming Kartogenin-Loaded Chitosan Hydrogel with Tunable Rheological Properties for Cartilage Tissue Engineering. Colloids Surfaces B Biointerfaces 2020, 192, 111059, doi:10.1016/j.colsurfb.2020.111059.
- Tian, L.; Singh, A.; Singh, A.V. Synthesis and Characterization of Pectin-Chitosan Conjugate for Biomedical Application. J. Biol. Macromol. 2020, 153, 533–538, doi:10.1016/j.ijbiomac.2020.02.313.
- Ali, A.; Ahmed, S. A Review on Chitosan and Its Nanocomposites in Drug Delivery. J. Biol. Macromol. 2018, 109, 273–286, doi:10.1016/J.IJBIOMAC.2017.12.078.
- Situ, W.; Xiang, T.; Liang, Y. Chitosan-Based Particles for Protection of Proteins during Storage and Oral Administration. J. Biol. Macromol. 2018, 117, 308–314, doi:10.1016/J.IJBIOMAC.2018.05.208.
- Bakshi, P.S.; Selvakumar, D.; Kadirvelu, K.; Kumar, N.S. Chitosan as an Environment Friendly Biomaterial – a Review on Recent Modifications and Applications. J. Biol. Macromol. 2020, 150, 1072–1083, doi:10.1016/j.ijbiomac.2019.10.113.
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An Update on Potential Biomedical and Pharmaceutical Applications. Drugs 2015, 13, 5156–5186, doi:10.3390/md13085156.
- Ahmed, T.A.; Aljaeid, B.M. Preparation, Characterization, and Potential Application of Chitosan, Chitosan Derivatives, and Chitosan Metal Nanoparticles in Pharmaceutical Drug Delivery. Drug Des. Devel. Ther. 2016, 10, 483–507, doi:10.2147/DDDT.S99651.
- Castro, P.M.; Raquel, A.; Sarmento, B.; Pintado, M. Recent Insights in the Use of Nanocarriers for the Oral Delivery of Bioactive Proteins and Peptides. Peptides 2018, doi:10.1016/j.peptides.2018.01.002.
- Rizvi, S.A.A.; Saleh, A.M. Applications of Nanoparticle Systems in Drug Delivery Technology. Saudi Pharm. J. 2017, 26, 64–70, doi:10.1016/j.jsps.2017.10.012.
- Luo, Y.Y.; Xiong, X.Y.; Tian, Y.; Li, Z.L.; Gong, Y.C.; Li, Y.P. A Review of Biodegradable Polymeric Systems for Oral Insulin Delivery. Drug Deliv. 2015, 23, 1882–1891, doi:10.3109/10717544.2015.1052863.
- Hu, Q.; Luo, Y. Recent Advances of Polysaccharide-Based Nanoparticles for Oral Insulin Delivery. Int. J. Biol. Macromol. 2018, 120, 775–782, doi:10.1016/j.ijbiomac.2018.08.152.
- Tavernini, L.; Ottone, C.; Illanes, A.; Wilson, L. Entrapment of Enzyme Aggregates in Chitosan Beads for Aroma Release in White Wines. Int. J. Biol. Macromol. 2020, 154, 1082–1090, doi:10.1016/j.ijbiomac.2020.03.031.
- Bugnicourt, L.; Ladavière, C. Interests of Chitosan Nanoparticles Ionically Cross-Linked with Tripolyphosphate for Biomedical Applications. Prog. Polym. Sci. 2016, 60, 1–17, doi:10.1016/j.progpolymsci.2016.06.002.
- Fernando, I.P.S.; Lee, W.W.; Han, E.J.; Ahn, G. Alginate-Based Nanomaterials: Fabrication Techniques, Properties, and Applications. Chem. Eng. J. 2019, 391, 123823, doi:10.1016/j.cej.2019.123823.
- Quiñones, J.P.; Peniche, H.; Peniche, C. Chitosan Based Self-Assembled Nanoparticles in Drug Delivery. Polymers (Basel) 2018, 10, 235, doi:10.3390/polym10030235.
- Mukhopadhyay, P.; Chakraborty, S.; Bhattacharya, S.; Mishra, R.; Kundu, P.P. PH-Sensitive Chitosan/Alginate Core-Shell Nanoparticles for Efficient and Safe Oral Insulin Delivery. Int. J. Biol. Macromol. 2015, 72, 640–648, doi:10.1016/J.IJBIOMAC.2014.08.040.
- Bhattacharyya, A.; Mukherjee, D.; Mishra, R.; Kundu, P.P. Preparation of Polyurethane–Alginate/Chitosan Core Shell Nanoparticles for the Purpose of Oral Insulin Delivery. Eur. Polym. J. 2017, 92, 294–313, doi:10.1016/j.eurpolymj.2017.05.015.
- Chen, T.; Li, S.; Zhu, W.; Liang, Z.; Zeng, Q. Self-Assembly PH-Sensitive Chitosan/Alginate Coated Polyelectrolyte Complexes for Oral Delivery of Insulin. J. Microencapsul. 2019, 36, 96–107, doi:10.1080/02652048.2019.1604846.
- Pereira De Sousa, I.; Moser, T.; Steiner, C.; Fichtl, B.; Bernkop-Schnürch, A. Insulin Loaded Mucus Permeating Nanoparticles: Addressing the Surface Characteristics as Feature to Improve Mucus Permeation. Int. J. Pharm. 2016, 500, 236–244, doi:10.1016/j.ijpharm.2016.01.022.
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Formulation and Characterisation of Insulin-Loaded Chitosan Nanoparticles Capable of Inducing Glucose Uptake in Skeletal Muscle Cells in Vitro. J. Drug Deliv. Sci. Technol. 2020, 57, 101738, doi:10.1016/j.jddst.2020.101738.
- Erel, G.; Kotmakçı, M.; Akbaba, H.; Sözer Karadağlı, S.; Kantarcı, A.G. Nanoencapsulated Chitosan Nanoparticles in Emulsion-Based Oral Delivery System: In Vitro and in Vivo Evaluation of Insulin Loaded Formulation. J. Drug Deliv. Sci. Technol. 2016, 36, 161–167, doi:10.1016/J.JDDST.2016.10.010.
- He, Z.; Santos, J.L.; Tian, H.; Huang, H.; Hu, Y.; Liu, L.; Leong, K.W.; Chen, Y.; Mao, H.-Q. Scalable Fabrication of Size-Controlled Chitosan Nanoparticles for Oral Delivery of Insulin. Biomaterials 2017, 130, 28–41, doi:10.1016/J.BIOMATERIALS.2017.03.028.
- Wiessner, J.H.; Hwang, K.J. Binding of Insulin to the External Surface of Liposomes. Effect of Surface Curvature, Temperature, and Lipid Composition. BBA Biomembr. 1982, 689, 490–498, doi:10.1016/0005-2736(82)90307-8.
- Al-Remawi, M.; Elsayed, A.; Maghrabi, I.; Hamaidi, M.; Jaber, N. Chitosan/Lecithin Liposomal Nanovesicles as an Oral Insulin Delivery System. Pharm. Dev. Technol. 2017, 22, 390–398, doi:10.1080/10837450.2016.1213745.
- Sahoo, P.; Leong, K.H.; Nyamathulla, S.; Onuki, Y.; Takayama, K.; Chung, L.Y. Optimization of PH-Responsive Carboxymethylated Iota-Carrageenan/Chitosan Nanoparticles for Oral Insulin Delivery Using Response Surface Methodology. React. Funct. Polym. 2017, 119, 145–155, doi:10.1016/j.reactfunctpolym.2017.08.014.