Factor XI (FXI) is one promising target. Existing direct oral anticoagulants (DOACs)DOACs have improved treatment options compared to the classic heparins and vitamin K antagonists (VKA) VKA, but the bleeding risks associated with their use are substantial enough to expand the focus onto the development of their antidotes. Early indications are that FXI-directed strategies could offer similar protection against thrombotic events as DOACs, but with the added benefit of lower bleeding risk. Furthermore, the spectrum of modalities for FXI inhibition presents a range of options in both types of administration and duration of effect. With the possibility of once- or twice-monthly injections, some FXI-directed agents could also improve treatment compliance compared to current therapies. Altogether, FXIa inhibitors could be a therapeutic option in a broad spectrum of clinical scenarios.
- factor XI
- factor XI inhibitor
- thrombosis
1. Introduction
2. Distinguishing Physiological Hemostasis from Pathological Thrombosis
Hemostasis is the normal, physiological process by which the clotting cascade seals up vascular damage to limit blood loss following injury. Thrombosis, on the other hand, encompasses various pathological conditions where the normally physiological clotting processes end up generating blood clot(s) inside the vascular lumen that are disruptive to the normal flow of blood. Thrombin generation and fibrin formation are the culminating steps in both hemostasis and thrombosis, but with important differences in the pathways involved. Hemostasis is commonly triggered when tissue factor (TF) within the adventitial layer of blood vessels gets exposed to blood. Injury to vasculature that can lead to bleeding activates a series of soluble plasma proteins that act together in a cascade of enzyme activation events and culminate in the formation of platelet-fibrin clot(s). Because of the relatively high concentration of TF in such scenarios, the generation of thrombin is rapid and intense, quickly forming a hemostatic plug that seals the inciting TF away from blood. This disrupts the amplification of the coagulation processes through feedback mechanisms to the point of becoming pathological. The concentration of TF in thrombosis is lower relative to hemostasis, but its duration of contact with blood components often lasts longer. Whether triggered by TF from disruption of an atherosclerotic plaque or activated monocytes/macrophages recruited to the site of injury or inflammation, or by implanted medical devices or neutrophil extracellular traps (NETs), these scenarios depend on the feedback mechanisms of the coagulation cascade for the growth and stabilization of the thrombus. This clot or thrombus can impede the flow of blood to the distal tissues and organs, leading to ischemia and necrosis, manifesting as clinical events including acute coronary syndrome, stroke, or deep vein thrombosis.3. Classic Coagulation Cascade
The two major pathways for triggering blood clotting cascade are well known; (1) the tissue factor pathway and (2) the contact pathway. Both pathways trigger a series of cascading events that generate a blood clot (Figure 1) with the purpose to separate and seal the triggering agent from blood, thereby preventing its further contact with plasma components and arresting the thrombotic process.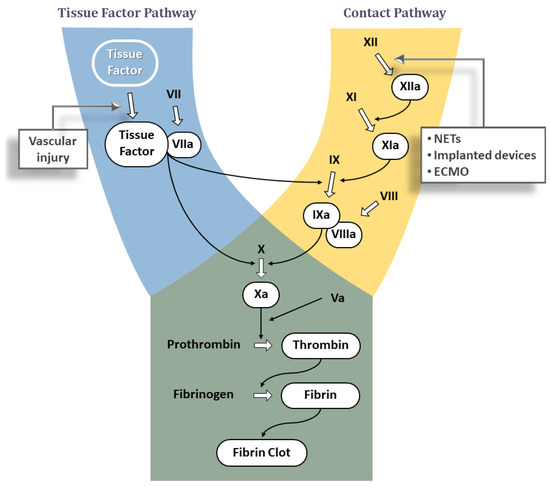
3.1. Tissue Factor Pathway
This pathway is also known as the ‘Extrinsic’ pathway, as it is triggered by plasma components coming into contact with an agent that is extrinsic to blood (i.e., TF). The contact may happen when TF, normally embedded in the vascular wall, is exposed to blood due to rupture of a plaque, or when TF is expressed on the surface of cells active in inflammatory and immunological processes (e.g., monocytes and macrophages). Tissue Factor is an integral cell-membrane protein that forms a complex with the coagulation factor VIIa (FVIIa), normally present in plasma in the inactive zymogen form FVII. The TF:FVIIa complex is a potent activator of coagulation and converts factors IX (FIX) and X (FX) to the active forms FIXa and FXa, respectively (Figure 1). Each of these active enzymes assembles with its protein cofactor (FVIIIa and FVa, respectively) on suitable membrane surfaces to further propagate the coagulation cascade. The end result is a large burst of thrombin, the last serine protease in the clotting cascade. Thrombin not only converts fibrinogen into fibrin via limited proteolysis—which in turn assembles into a fibrin clot—but is also one of the most potent activators of platelets. The activation and aggregation of platelets contributes to the formation of a hemostatic plug. Additionally, thrombin also activates FV, FVIII, and FXI, the latter two of which are part of the contact pathway. Thus, the initial thrombin generated by the TF pathway can lead to activation of the contact pathway.3.2. Contact Pathway
Also known as the ‘Intrinsic’ pathway, this pathway is triggered when blood comes into contact with anionic surfaces, such as extracellular DNA, RNA from activated or dying cells including neutrophil extracellular traps (NETs) released by activated neutrophils [15], or polyphosphates from the dense granules of activated platelets or microorganisms [16], or those on artificial surfaces [17]. This leads to a change in the conformation of plasma factor XII (FXII) into the active factor XII (FXIIa) [18][19]. FXIIa activates Prekallikrein to Kallikrein, which in turn reciprocally activates FXII to FXIIa in a positive feedback loop [20]. Downstream, FXIIa activates FXI to FXIa, which in turn leads to proteolysis of factor IX (FIX) to the active form (FIXa). The complex of FIXa and FVIIIa then activates FX to FXa at the point where the TF and contact pathways converge to form the final common pathway (Figure 1). The end result of all these interactions again is thrombin generation and formation of a blood clot.
4. Factor XI as a Therapeutic Target
Factor XI is a blood coagulation zymogen produced by the liver that is part of the early phase of the contact pathway [21]. It is converted to the active serine protease FXIa by thrombin, FXIIa, and by FXIa itself and in turn activates FIX to further advance the coagulation process [21]. FXI plays an important part in blood coagulation because its feedback activation amplifies in vivo thrombin generation and fibrin formation [22]. The additional thrombin formed via the FXI feedback loop also promotes the activation of Thrombin Activatable Fibrinolysis Inhibitor (TAFI), which increases the clot’s resistance to fibrinolysis, thereby helping to stabilize the formed clot. The greater role of FXIa in thrombosis compared to hemostasis is evident from several epidemiological and genetic studies. Higher levels of circulating FXI levels are associated with increased risk for venous and arterial thrombosis, including stroke [23][24]. Deficiency of FXI (Hemophilia C, Plasma Thromboplastin Antecedent Deficiency, Rosenthal Syndrome) is rare and characterized by little to no bleeding tendency. Bleeding risk with factor XI deficiency selectively increases in tissues with high fibrinolytic activity (e.g., following dental surgery, tonsillectomy, and prostate surgery) [25]. Most frequent presentations involve nosebleeds or bleeding after tooth extractions. In fact, patients suffering from congenital FXI deficiency appear to have some degree of protection from thrombotic events, with lower rates of ischemic stroke and venous thromboembolism [26][27].5. Pharmacologic Strategies for Factor XI Inhibition
Given the larger role FXI is thought to play in thrombosis than in hemostasis, novel approaches to inhibit its generation and activity are being explored as new therapeutic strategies (Figure 23). These include: (a) Antisense Oligonucleotides (ASOs) that act on the liver to knockdown hepatic synthesis of FXI, (b) small molecules that target the FXI active site or the heparin allosteric site on FXIa, (c) monoclonal antibodies that act by blocking the activation or inhibiting the activity, and (d) Aptamers.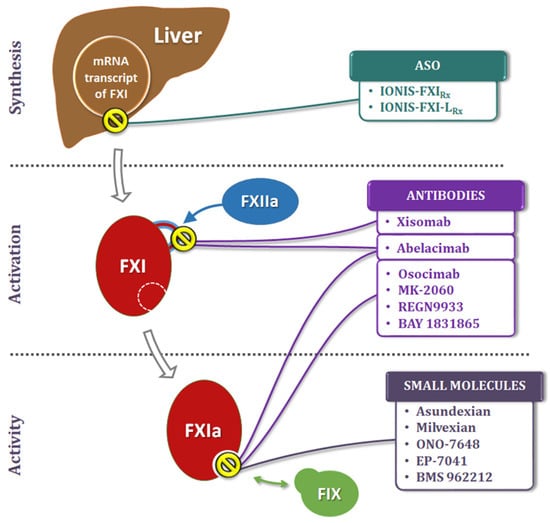
5.1. Antisense Oligonucleotides (ASOs)
Antisense Oligonucleotides are short, single-stranded nucleic acid sequences that pair with specific regions of mRNA and regulate its gene expression [28][29], thereby downgrading the hepatic synthesis of FXI. Their benefits include high specificity, predictable pharmacokinetics (PK), and long half-life. Furthermore, ASOs lack the drug–drug interactions commonly seen with conventional therapeutic agents.-
IONIS-FXIRx.
-
IONIS-FXI-LRx.
5.2. Small Molecules
5.2.1. Small Molecules Targeting the Active Site on FXIa
-
Asundexian (BAY 2433334).
-
Milvexian (JNJ-70033093/BMS-986177).
-
Other Small-Molecules in Early Development
5.2.2. Small Molecules Targeting Heparin Allosteric Site on FXIa
This group of FXIa-directed agents exert their inhibitory effects by attaching to the heparin-binding site on the catalytic domain of FXIa. Given the structural similarities between the active sites of various serine proteases, it is believed that allosteric inhibition would have the advantage of being more specific. Some of the sulfated glycosaminoglycan (SPGG) mimetic compounds under development in this group not only exhibit a highly selective inhibition of FXIa than any other target in the coagulation cascade, but also display a reversal of their anticoagulant effects with FXI and serum albumin [37].
5.3. Monoclonal Antibodies
-
Abelacimab (MAA868).
-
Osocimab (BAY 1213790).
-
Xisomab 3G3 (AB023).
-
Other Antibodies in Clinical Testing.
5.4. Aptamers
Aptamers are single-stranded oligonucleotides that act as potent antagonists by binding to their target protein. A number of specific aptamers have been developed that serve as strong anticoagulants by disrupting complex interactions on their target proteins [41]. To date, aptamers targeting FXI directly or indirectly are in very early stages, with none reaching clinical development. In laboratory testing, an agent designated Factor ELeven Inhibitory APtamer (FELIAP) was shown to competitively inhibit FXIa-catalyzed FIX activation and complex formation with antithrombin, without affecting FXI activation itself. Plasma clotting and thrombin generation assays were also inhibited by this aptamer [42].6. Therapeutic Investigation
6.1. Active Areas of Investigations
6.1.1. Atrial Fibrillation
It is the most common clinically significant arrhythmia [43], with an age-related risk of occurrence, and cardiac thrombus formation and systemic embolization are its most significant clinical complications, raising the risk of stroke by 4–5 fold [44][45]. The DOACs have shown better results than warfarin in preventing stroke in non-valvular AF patients, with lower or equivalent rates of bleeding complications [46]. However, the need for safer agents still persists and is even more pressing in AF patients requiring hemodialysis. There is uncertainty as to whether the benefits of VKA actually outweigh their harm in AF patients requiring hemodialysis, and trials investigating the role of DOACs in this population are mostly in the early stages. Even in the absence of AF, hemodialysis on its own is a major problem, with cardiovascular events accounting for nearly half of the mortality in these patients. The availability of a newer antithrombotic agent with a better safety profile than existing strategies could significantly improve clinical outcomes in AF patients with or without the need for hemodialysis and in those who require dialysis, with or without AF. An FXI-inhibiting strategy could be an improved therapeutic option in these patients and warrants investigation in clinical trials.6.1.2. Venous Thromboembolism
Anticoagulant therapy is the mainstay for the prevention and treatment of VTE diseases. The development of DOACs has improved the management of VTE compared to where it was with LHMH/VKA [47]. As a result, rates of idiopathic VTE appear to be on the decline, but the incidence of non-idiopathic DVT and PE seem to be steady or increasing [48], highlighting the need for newer treatment options. Even when used at reduced doses, there is a risk of bleeding with DOAC therapy in these patients [49].6.2. Potential Areas for Therapeutic Investigations
Inhibitors of FXI/FXIa are currently in the early stages of clinical development, and over time the spectrum of their clinical application will evolve into specific, focused indications. The areas for the investigation of their therapeutic applications potentially include any pathology where thromboembolism plays an important role. Given the wide-ranging times of their onset and duration of action, FXIa inhibitors have the potential to develop into therapeutic strategies for the treatment and prevention of both acute and chronic, venous, and arterial thromboembolic disorders.
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128.
- Day, I.S.C.f.W.T. Thrombosis: A major contributor to the global disease burden. J. Thromb. Haemost. 2014, 12, 1580–1590.
- O’Brien, M.P.; Zafar, M.U.; Rodriguez, J.C.; Okoroafor, I.; Heyison, A.; Cavanagh, K.; Rodriguez-Caprio, G.; Weinberg, A.; Escolar, G.; Aberg, J.A.; et al. Targeting thrombogenicity and inflammation in chronic HIV infection. Sci. Adv. 2019, 5, eaav5463.
- Hemkens, L.G.; Bucher, H.C. HIV infection and cardiovascular disease. Eur. Heart J. 2014, 35, 1373–1381.
- Alonso, A.; Barnes, A.E.; Guest, J.L.; Shah, A.; Shao, I.Y.; Marconi, V. HIV Infection and Incidence of Cardiovascular Diseases: An Analysis of a Large Healthcare Database. J. Am. Heart Assoc. 2019, 8, e012241.
- Garcia, D.A.; Baglin, T.P.; Weitz, J.I.; Samama, M.M. Parenteral anticoagulants: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e24S–e43S.
- Ageno, W.; Gallus, A.S.; Wittkowsky, A.; Crowther, M.; Hylek, E.M.; Palareti, G. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e44S–e88S.
- Zafar, M.U.; Vorchheimer, D.A.; Gaztanaga, J.; Velez, M.; Yadegar, D.; Moreno, P.R.; Kunitada, S.; Pagan, J.; Fuster, V.; Badimon, J.J. Antithrombotic effects of factor Xa inhibition with DU-176b: Phase-I study of an oral, direct factor Xa inhibitor using an ex-vivo flow chamber. Thromb. Haemost. 2007, 98, 883–888.
- Zafar, M.U.; Farkouh, M.E.; Osende, J.; Shimbo, D.; Palencia, S.; Crook, J.; Leadley, R.; Fuster, V.; Chesebro, J.H. Potent arterial antithrombotic effect of direct factor-Xa inhibition with ZK-807834 administered to coronary artery disease patients. Thromb. Haemost. 2007, 97, 487–492.
- Viles-Gonzalez, J.F.; Gaztanaga, J.; Zafar, U.M.; Fuster, V.; Badimon, J.J. Clinical and experimental experience with factor Xa inhibitors. Am. J. Cardiovasc. Drugs 2004, 4, 379–384.
- Sardar, P.; Chatterjee, S.; Lavie, C.J.; Giri, J.S.; Ghosh, J.; Mukherjee, D.; Lip, G.Y. Risk of major bleeding in different indications for new oral anticoagulants: Insights from a meta-analysis of approved dosages from 50 randomized trials. Int. J. Cardiol. 2015, 179, 279–287.
- Hellenbart, E.L.; Faulkenberg, K.D.; Finks, S.W. Evaluation of bleeding in patients receiving direct oral anticoagulants. Vasc. Health Risk Manag. 2017, 13, 325–342.
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962.
- Alamneh, E.A.; Chalmers, L.; Bereznicki, L.R. Suboptimal Use of Oral Anticoagulants in Atrial Fibrillation: Has the Introduction of Direct Oral Anticoagulants Improved Prescribing Practices? Am. J. Cardiovasc. Drugs 2016, 16, 183–200.
- Sorvillo, N.; Cherpokova, D.; Martinod, K.; Wagner, D.D. Extracellular DNA NET-Works With Dire Consequences for Health. Circ. Res. 2019, 125, 470–488.
- Baker, C.J.; Smith, S.A.; Morrissey, J.H. Polyphosphate in thrombosis, hemostasis, and inflammation. Res. Pract. Thromb. Haemost. 2019, 3, 18–25.
- Nossel, H.L. Differential consumption of coagulation factors resulting from activation of the extrinsic (tissue thromboplastin) or the intrinsic (foreign surface contact) pathways. Blood 1967, 29, 331–340.
- Silverberg, M.; Dunn, J.T.; Garen, L.; Kaplan, A.P. Autoactivation of human Hageman factor. Demonstration utilizing a synthetic substrate. J. Biol. Chem. 1980, 255, 7281–7286.
- Tankersley, D.L.; Finlayson, J.S. Kinetics of activation and autoactivation of human factor XII. Biochemistry 1984, 23, 273–279.
- Franke, L.; Schewe, H.J.; Muller, B.; Campman, V.; Kitzrow, W.; Uebelhack, R.; Berghofer, A.; Muller-Oerlinghausen, B. Serotonergic platelet variables in unmedicated patients suffering from major depression and healthy subjects: Relationship between 5HT content and 5HT uptake. Life Sci. 2000, 67, 301–315.
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370.
- Vondemborne, P.A.K.; Meijers, J.C.M.; Bouma, B.N. Feedback Activation of Factor-Xi by Thrombin in Plasma Results in Additional Formation of Thrombin That Protects Fibrin Clots from Fibrinolysis. Blood 1995, 86, 3035–3042.
- Meijers, J.C.; Tekelenburg, W.L.; Bouma, B.N.; Bertina, R.M.; Rosendaal, F.R. High levels of coagulation factor XI as a risk factor for venous thrombosis. N. Engl. J. Med. 2000, 342, 696–701.
- Gill, D.; Georgakis, M.K.; Laffan, M.; Sabater-Lleal, M.; Malik, R.; Tzoulaki, I.; Veltkamp, R.; Dehghan, A. Genetically Determined FXI (Factor XI) Levels and Risk of Stroke. Stroke J. Cereb. Circ. 2018, 49, 2761–2763.
- Salomon, O.; Steinberg, D.M.; Seligshon, U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia 2006, 12, 490–493.
- Salomon, O.; Steinberg, D.M.; Koren-Morag, N.; Tanne, D.; Seligsohn, U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood 2008, 111, 4113–4117.
- Preis, M.; Hirsch, J.; Kotler, A.; Zoabi, A.; Stein, N.; Rennert, G.; Saliba, W. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood 2017, 129, 1210–1215.
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140.
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600.
- Younis, H.S.; Crosby, J.; Huh, J.I.; Lee, H.S.; Rime, S.; Monia, B.; Henry, S.P. Antisense inhibition of coagulation factor XI prolongs APTT without increased bleeding risk in cynomolgus monkeys. Blood 2012, 119, 2401–2408.
- Buller, H.R.; Bethune, C.; Bhanot, S.; Gailani, D.; Monia, B.P.; Raskob, G.E.; Segers, A.; Verhamme, P.; Weitz, J.I.; Investigators, F.-A.T. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N. Engl. J. Med. 2015, 372, 232–240.
- Rao, S.V.; Kirsch, B.; Bhatt, D.L.; Budaj, A.; Coppolecchia, R.; Eikelboom, J.; James, S.K.; Jones, W.S.; Merkely, B.; Keller, L.; et al. A Multicenter, Phase 2, Randomized, Placebo-Controlled, Double-Blind, Parallel-Group, Dose-Finding Trial of the Oral Factor XIa Inhibitor Asundexian to Prevent Adverse Cardiovascular Outcomes Following Acute Myocardial Infarction. Circulation 2022, 146, 1196–1206.
- Weitz, J.I.; Strony, J.; Ageno, W.; Gailani, D.; Hylek, E.M.; Lassen, M.R.; Mahaffey, K.W.; Notani, R.S.; Roberts, R.; Segers, A.; et al. Milvexian for the Prevention of Venous Thromboembolism. N. Engl. J. Med. 2021, 385, 2161–2172.
- Beale, D.; Dennison, J.; Boyce, M.; Mazzo, F.; Honda, N.; Smith, P.; Bruce, M. ONO-7684 a novel oral FXIa inhibitor: Safety, tolerability, pharmacokinetics and pharmacodynamics in a first-in-human study. Br. J. Clin. Pharm. 2021, 87, 3177–3189.
- Hayward, N.J.; Goldberg, D.I.; Morrel, E.M.; Friden, P.M.; Bokesch, P.M. Phase 1a/1b Study of EP-7041: A Novel, Potent, Selective, Small Molecule FXIa Inhibitor. Circulation 2017, 136, 13747.
- Perera, V.; Luettgen, J.M.; Wang, Z.; Frost, C.E.; Yones, C.; Russo, C.; Lee, J.; Zhao, Y.; LaCreta, F.P.; Ma, X.; et al. First-in-human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS-962212, a direct, reversible, small molecule factor XIa inhibitor in non-Japanese and Japanese healthy subjects. Br. J. Clin. Pharm. 2018, 84, 876–887.
- Al-Horani, R.A.; Gailani, D.; Desai, U.R. Allosteric inhibition of factor XIa. Sulfated non-saccharide glycosaminoglycan mimetics as promising anticoagulants. Thromb. Res. 2015, 136, 379–387.
- Koch, A.W.; Schiering, N.; Melkko, S.; Ewert, S.; Salter, J.; Zhang, Y.; McCormack, P.; Yu, J.; Huang, X.; Chiu, Y.H.; et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood 2019, 133, 1507–1516.
- Weitz, J.I.; Bauersachs, R.; Becker, B.; Berkowitz, S.D.; Freitas, M.C.S.; Lassen, M.R.; Metzig, C.; Raskob, G.E. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. JAMA 2020, 323, 130–139.
- Lorentz, C.U.; Tucker, E.I.; Verbout, N.G.; Shatzel, J.J.; Olson, S.R.; Markway, B.D.; Wallisch, M.; Ralle, M.; Hinds, M.T.; McCarty, O.J.T.; et al. The contact activation inhibitor AB023 in heparin-free hemodialysis: Results of a randomized phase 2 clinical trial. Blood 2021, 138, 2173–2184.
- White, R.R.; Sullenger, B.A.; Rusconi, C.P. Developing aptamers into therapeutics. J. Clin. Investig. 2000, 106, 929–934.
- Donkor, D.A.; Bhakta, V.; Eltringham-Smith, L.J.; Stafford, A.R.; Weitz, J.I.; Sheffield, W.P. Selection and characterization of a DNA aptamer inhibiting coagulation factor XIa. Sci. Rep. 2017, 7, 2102.
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375.
- Wolf, P.A.; Abbott, R.D.; Kannel, W.B. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke J. Cereb. Circ. 1991, 22, 983–988.
- Kannel, W.B.; Benjamin, E.J. Status of the epidemiology of atrial fibrillation. Med. Clin. N. Am. 2008, 92, 17–40.
- Lopez-Lopez, J.A.; Sterne, J.A.C.; Thom, H.H.Z.; Higgins, J.P.T.; Hingorani, A.D.; Okoli, G.N.; Davies, P.A.; Bodalia, P.N.; Bryden, P.A.; Welton, N.J.; et al. Oral anticoagulants for prevention of stroke in atrial fibrillation: Systematic review, network meta-analysis, and cost effectiveness analysis. BMJ Clin. Res. Ed 2017, 359, j5058.
- van Es, N.; Coppens, M.; Schulman, S.; Middeldorp, S.; Buller, H.R. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: Evidence from phase 3 trials. Blood 2014, 124, 1968–1975.
- Heit, J.A.; Petterson, T.M.; Farmer, S.A.; Bailey, K.R.; Joseph Melton, L. Trends in the incidence of deep vein thrombosis and pulmonary embolism: A 35-year population-based study. Blood 2006, 108, 1488.
- van der Hulle, T.; Kooiman, J.; den Exter, P.L.; Dekkers, O.M.; Klok, F.A.; Huisman, M.V. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: A systematic review and meta-analysis. J. Thromb. Haemost. 2014, 12, 320–328.